API¶
ScTriangulate Class Methods¶
__init__()¶
- class sctriangulate.main_class.ScTriangulate(dir, adata, query, species='human', criterion=2, verbose=1, reference=None, add_metrics={'tfidf5': <function tf_idf5_for_cluster>}, predict_doublet=False)[source]¶
How to create/instantiate ScTriangulate object.
- Parameters
dir – Output folder path on the disk, will create if not exist
adata – input adata file
query – a python list contains the annotation names to query
species – string, either human (default) or mouse, it will impact how the program searches for artifact genes in the database
criterion –
int, it controls what genes would be considered as artifact genes:
criterion1: all will be artifact
criterion2: all will be artifact except cellcycle [Default]
criterion3: all will be artifact except cellcycle, ribosome
criterion4: all will be artifact except cellcycle, ribosome, mitochondrial
criterion5: all will be artifact except cellcycle, ribosome, mitochondrial, antisense
criterion6: all will be artifact except cellcycle, ribosome, mitochondrial, antisense, predict_gene
verbose – int, it controls how the log file will be generated. 1 means print to stdout (default), 2 means print to a file in the directory specified by dir parameter.
add_metrics – python dictionary. These allows users to add additional metrics to favor or disqualify certain cluster. By default, we add tfidf5 score {‘tfidf5’:tf_idf5_for_cluster}, remember the value in the dictionary should be the name of a callable, user can define the callable by themselves. If don’t want any addded metrics, using empty dict {}.
predict_doublet –
boolean or string, whether to predict doublet using scrublet or not. Valid value:
True: will predict doublet score
False: will not predict doublet score
(string) precomputed: will not predict doublet score but just use existing one
Note
For the callable, the signature should be func(adata,key,**kwargs) -> mapping {cluster1:0.5,cluster2:0.6}, when running the program in lazy_run function, we need to specify added_metrics_kwargs as a list, each element in the list is a dictionary that corresponds to the kwargs that will be passed to each callable.
Example:
adata = sc.read('pbmc3k_azimuth_umap.h5ad') sctri = ScTriangulate(dir='./output',adata=adata,query=['leiden1','leiden2','leiden3'])
(statis) deserialize()¶
- sctriangulate.main_class.ScTriangulate.deserialize(name)¶
This is static method, to deserialize a pickle file on the disk back to the ram as a sctri object
- Parameters
name – string, the name of the pickle file on the disk.
Examples:
ScTriangulate.deserialize(name='after_rank_pruning.p')
(statics) salvage_run()¶
- sctriangulate.main_class.ScTriangulate.salvage_run(step_to_start, last_step_file, outdir=None, scale_sccaf=True, layer=None, added_metrics_kwargs=[{'species': 'human', 'criterion': 2, 'layer': None}], compute_shapley_parallel=True, shapley_mode='shapley_all_or_none', shapley_bonus=0.01, win_fraction_cutoff=0.25, reassign_abs_thresh=10, assess_raw=False, assess_pruned=True, viewer_cluster=True, viewer_cluster_keys=None, viewer_heterogeneity=True, viewer_heterogeneity_keys=None, nca_embed=False, n_top_genes=3000, other_umap=None, heatmap_scale=None, heatmap_cmap='viridis', heatmap_regex=None, heatmap_direction='include', heatmap_n_genes=None, heatmap_cbar_scale=None)¶
This is a static method, which allows to user to resume running scTriangulate from certain point, instead of running from very beginning if the intermediate files are present and intact.
- Parameters
step_to_start – string, now support ‘assess_pruned’.
last_step_file – string, the path to the intermediate from which we start the salvage run.
outdir – None or string, whether to change the outdir or not.
Other parameters are the same as
lazy_runfunction.Examples:
ScTriangulate.salvage_run(step_to_start='assess_pruned',last_step_file='output/after_rank_pruning.p')
add_new_metrics()¶
- sctriangulate.main_class.ScTriangulate.add_new_metrics(self, add_metrics)¶
Users can add new callable or pre-implemented function to the sctri.metrics attribute.
- Parameters
add_metrics – dictionary like {‘metric_name’: callable}, the callable can be a string of a scTriangulate pre-implemented function, for example, ‘tfidf5’,’tfidf1’. Or a callable.
Examples:
sctri.add_new_metrics(add_metrics={'tfidf1':tfidf1}) # make sure first from sctriangualte.metrics import tfidf1
add_to_invalid()¶
- sctriangulate.main_class.ScTriangulate.add_to_invalid(self, invalid)¶
add individual raw cluster names to the sctri.invalid attribute list.
- Parameters
invalid – list or string, contains the raw cluster names to add
Examples:
sctri.add_to_invalid(invalid=['annotation1@c3','annotation2@4']) sctri.add_to_invalid(invalid='annotation1@3')
add_to_invalid_by_win_fraction()¶
- sctriangulate.main_class.ScTriangulate.add_to_invalid_by_win_fraction(self, percent=0.25)¶
add individual raw cluster names to the sctri.invalid attribute list by win_fraction
- Parameters
percent – float, from 0-1, the fraction of cells within a cluster that were kept after the game. Default: 0.25
Examples:
sctri.add_to_invalid_by_win_fraction(percent=0.25)
clear_invalid()¶
- sctriangulate.main_class.ScTriangulate.clear_invalid(self)¶
reset/clear the sctri.invalid to an empty list
Examples:
sctri.clear_invalid()
cluster_performance()¶
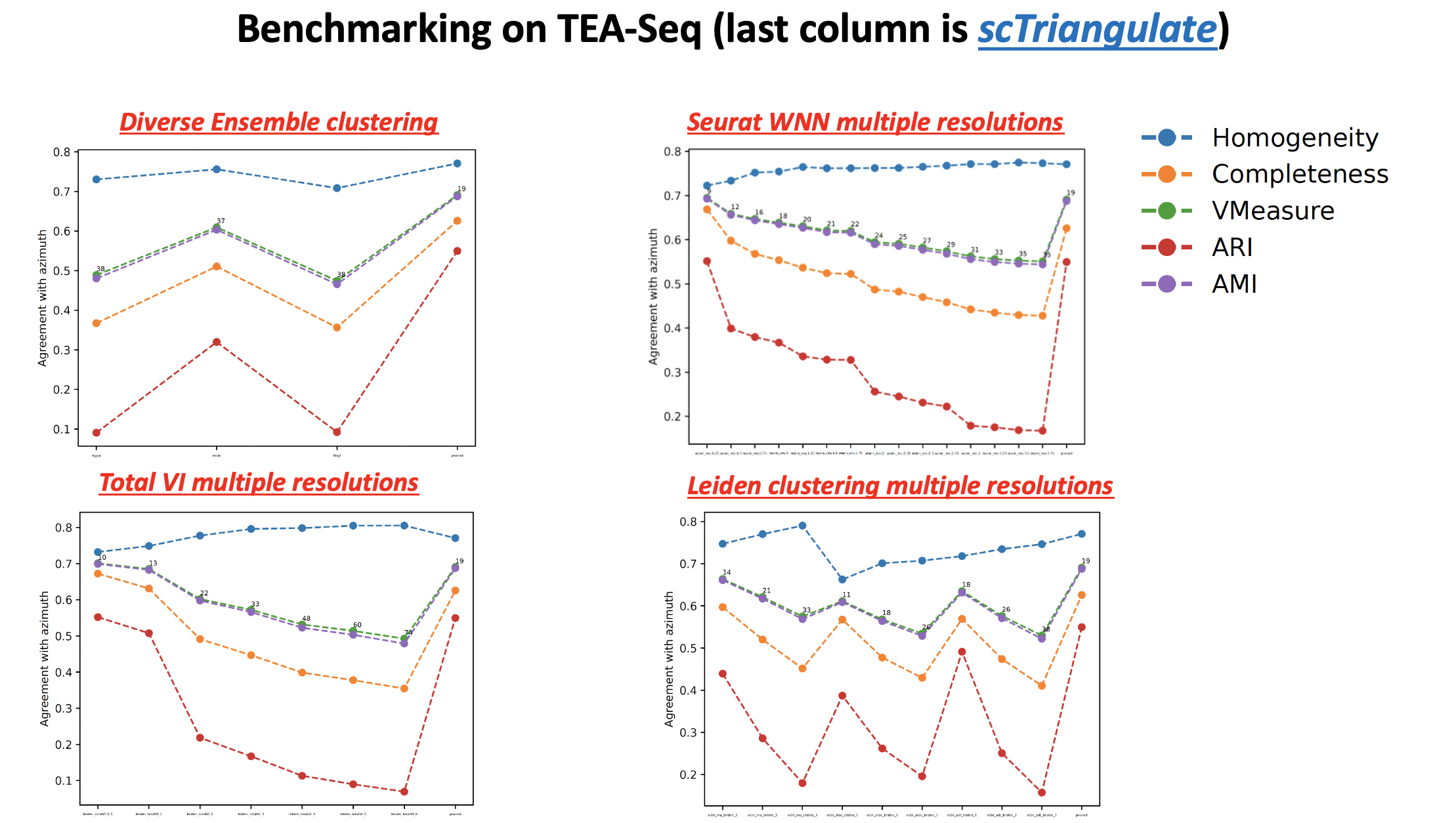
- sctriangulate.main_class.ScTriangulate.cluster_performance(self, cluster, competitors, reference, show_cluster_number=False, metrics=None, ylim=None, save=True, format='pdf')¶
automatic benchmark of scTriangulate clusters with all the individual or competitor annotation, against a ‘gold standard’ annotation, measured by all unsupervised cluster metrics (homogeneity, completeness, v_measure, ARI, NMI).
- Parameters
cluster – string, the scTriangulate annotation column name, for example, pruned
competitiors – list of string, each is a column name of a competitor annotation
reference – string, the column name containing reference annotation, for example, azimuth
show_cluster_number – bool, whether to show the number of cluster of each annotation in the performance line plot
metrics – None or any other, if not None, ARI and NMI will also be plotted
ylim – None or a tuple, specifiying the ylims of plot
save – bool, whether to save the figure
format – string, default is pdf, the format to save
Examples:
sctri.cluster_performance(cluster='pruned',competitors=['sctri_rna_leiden_1','sctri_rna_leiden_2','sctri_rna_leiden_3'], reference='azimuth',show_cluster_number=True,metrics=None)
compute_metrics()¶
- sctriangulate.main_class.ScTriangulate.compute_metrics(self, parallel=True, scale_sccaf=False, layer=None, added_metrics_kwargs=[{'species': 'human', 'criterion': 2, 'layer': None}], cores=None)¶
main function for computing the metrics (defined by self.metrics) of each clusters in each annotation. After the run, q (# query) * m (# metrics) columns will be added to the adata.obs, the column like will be like {metric_name}@{query_annotation_name}, i.e. reassign@sctri_rna_leiden_1
- Parameters
parallel – boolean, whether to run in parallel. Since computing metrics for each query annotation is idependent, the program will automatically employ q (# query) cores under the hood. If you want to fully leverage this feature, please make sure you specify at least q (# query) cores when running the program. It is highly recommend to run this in parallel. However, when the dataset is super large and have > 10 query annotation, we may encounter RAM overhead, in this case, sequential mode will be needed. Default: True
scale_sccaf – boolean, when running SCCAF score, since it is a logistic regression problem at its core, this parameter controls whether scale the expression data or not. It is recommended to scale the data for any machine learning algorithm, however, the liblinaer solver has been demonstrated to be robust to the scale/unscale options. When the dataset > 50,000 cells or features > 1000000 (ATAC peaks), it is advised to not scale it for faster running time.
layer – None or str, the adata layer where the raw count is stored, useful when calculating tfidf score when adata.X has been skewed (no zero value, like totalVI denoised value)
added_metrics_kwargs – list, see the notes in __init__ function, this is to specify additional arguments that will be passed to each added metrics callable.
cores – None or int, how many cores you’d like to specify, by default, it is min(n_annotations,n_available_cores) for metrics computing, and n_available_cores for other parallelizable operations
Examples:
sctri.compute_metrics(parallel=False)
compute_shapley()¶
- sctriangulate.main_class.ScTriangulate.compute_shapley(self, parallel=True, mode='shapley_all_or_none', bonus=0.01, cores=None)¶
Main core function, after obtaining the metrics for each cluster. For each single cell, let’s calculate the shapley value for each annotation and assign the cluster to the one with highest shapley value.
- Parameters
parallel – boolean. Whether to run it in parallel. (scatter and gather). Default: True
mode –
string, accepted values:
shapley_all_or_none: default, computing shapley, and players only get points when it beats all
shapley: computing shapley, but players get points based on explicit ranking, say 3 players, if ranked first, you get 3, if running up, you get 2
rank_all_or_none: no shapley computing, importance based on ranking, and players only get points when it beats all
rank: no shapley computing, importance based on ranking, but players get points based on explicit ranking as described above
bonus – float, default is 0.01, an offset value so that if the runner up is just {bonus} inferior to first place, it will still be a valid cluster
cores – None or int, None will run mp.cpu_counts() to get all available cpus.
Examples:
sctri.compute_shapley(parallel=True)
confusion_to_df()¶
- sctriangulate.main_class.ScTriangulate.confusion_to_df(self, mode, key)¶
Print out the confusion matrix with cluster labels (dataframe).
- Parameters
mode – either ‘confusion_reassign’ or ‘confusion_sccaf’
mode – python string, for example, ‘annotation1’
Examples:
sctri.confusion_to_df(mode='confusion_reassign',key='annotation1')
display_hierarchy()¶
- sctriangulate.main_class.ScTriangulate.display_hierarchy(self, ref_col, query_col, save=True)¶
Display the hierarchy of suggestive sub-clusterings, see the example results down the page.
- Parameters
ref_col – string, the annotation/column name in adata.obs which we want to inspect how it can be sub-divided
query_col – string, any cluster annotation column name
save – boolean, whether to save it to a file or stdout. Default: True
Examples:
sctri.display_hierarchy(ref_col='sctri_rna_leiden_1',query_col='raw')

doublet_predict()¶
- sctriangulate.main_class.ScTriangulate.doublet_predict(self)¶
wrapper function of running scrublet, will add a column on adata.obs called ‘doublet_scores’
Examples:
sctri.doublet_predict()
elo_rating_like()¶
- sctriangulate.main_class.ScTriangulate.elo_rating_like(self)¶
Computing an overall quality score for each annotation, like the idea of
Elo Ratingin chess in which it can reflect the probability that one player will win in a chess match. Here, we argue the overall quality score should be defined by the average of all cells’ shapley value in all clusters, normalized by the number of players (annotation), and further normalized by number of clusters, as our computation of shapley is an additive process such that more player will result in higher shapley value. This step should be run after shapley value were evaluated.- Return result_dic
a dictionary keyed by annotation, value is overall quality score
Examples:
result_dic = sctri.elo_rating_like() # {'sctri_rna_leiden_1': 1.5053613872472829, 'sctri_rna_leiden_2': 1.0973714905049967, 'sctri_rna_leiden_3': 1.1032324231884296}
extract_stability()¶
- sctriangulate.main_class.ScTriangulate.extract_stability(self, keys=None)¶
To extract cluster stability information
- Params keys
a list, containing the annotation column names, None means all in self.query
Examples:
sctri.extract_stability(keys=['annotation1','annotation2'])
gene_to_df()¶
- sctriangulate.main_class.ScTriangulate.gene_to_df(self, mode, key, raw=False, col='purify', n=100)¶
Output {mode} genes for all clusters in one annotation (key), mode can be either ‘marker_genes’ or ‘exclusive_genes’.
- Parameters
mode – python string, either ‘marker_genes’ or ‘exclusive_genes’
key – python string, annotation name
raw – False will generate non-raw (human readable) format. Default: False
col – Only when mode==’marker_genes’, whether output ‘whole’ column or ‘purify’ column. Default: purify
n – Only when mode==’exclusive_genes’, how many top exclusively expressed genes will be printed for each cluster.
Examples:
sctri.gene_to_df(mode='marker_genes',key='annotation1') sctri.gene_to_df(mode='exclusive_genes',key='annotation1')
get_metrics_and_shapley()¶
- sctriangulate.main_class.ScTriangulate.get_metrics_and_shapley(self, barcode, save=True)¶
For one single cell, given barcode/or other unique index, generate the all conflicting cluster from each annotation, along with the metrics associated with each cluster, including shapley value.
- Parameters
barcode – string, the barcode for the cell you want to query.
save – save the returned dataframe to directory or not. Default: True
- Returns
DataFrame
Examples:
sctri.get_metrics_and_shapley(barcode='AAACCCACATCCAATG-1',save=True)
lazy_run()¶
- sctriangulate.main_class.ScTriangulate.lazy_run(self, compute_metrics_parallel=True, scale_sccaf=False, layer=None, cores=None, added_metrics_kwargs=[{'species': 'human', 'criterion': 2, 'layer': None}], compute_shapley_parallel=True, shapley_mode=None, shapley_bonus=0.01, win_fraction_cutoff=0.25, reassign_abs_thresh=10, assess_raw=False, assess_pruned=False, viewer_cluster=False, viewer_cluster_keys=None, viewer_heterogeneity=False, viewer_heterogeneity_keys=None, nca_embed=False, n_top_genes=3000, other_umap=None, heatmap_scale=None, heatmap_cmap='viridis', heatmap_regex=None, heatmap_direction='include', heatmap_n_genes=None, heatmap_cbar_scale=None)¶
This is the highest level wrapper function for running every step in one goal.
- Parameters
compute_metrics_parallel – boolean, whether to parallelize
compute_metricsstep. Default: Truescale_sccaf – boolean, whether to first scale the expression matrix before running sccaf score. Default: False
layer – None or str, the adata layer where the raw count is stored, useful when calculating tfidf score when adata.X has been skewed (no zero value, like totalVI denoised value)
cores – None or int, how many cores you’d like to specify, by default, it is min(n_annotations,n_available_cores) for metrics computing, and n_available_cores for other parallelizable operations
added_metrics_kwargs – list, see the notes in __init__ function, this is to specify additional arguments that will be passed to each added metrics callable.
compute_shapley_parallel – boolean, whether to parallelize
compute_parallelstep. Default: Trueshapley_mode –
string, accepted values:
shapley_all_or_none: computing shapley, and players only get points when it beats all
shapley: computing shapley, but players get points based on explicit ranking, say 3 players, if ranked first, you get 3, if running up, you get 2
rank_all_or_none: no shapley computing, importance based on ranking, and players only get points when it beats all
rank: no shapley computing, importance based on ranking, but players get points based on explicit ranking as described above
None: if n_annotations <= 15, use shapley_all_or_none, if n_anntations > 15, use rank
shapley_bonus – float, default is 0.01, an offset value so that if the runner up is just {bonus} inferior to first place, it will still be a valid cluster
win_fraction_cutoff – float, between 0-1, the cutoff for function
add_invalid_by_win_fraction. Default: 0.25reassign_abs_thresh – int, the cutoff for minimum number of cells a valid cluster should haves. Default: 10
assess_raw – boolean, whether to run the same cluster assessment metrics on raw cluster labels. Default: False
assess_pruned – boolean, whether to run same cluster assessment metrics on final pruned cluster labels. Default: False
viewer_cluster – boolean, whether to build viewer html page for all clusters’ diagnostic information. Default: False
viewer_cluster_keys – list, clusters from what annotations we want to view on the viewer, only clusters within this annotation whose diagnostic plot will be generated under the dir name figure4viewer. Default: None, means all annotations in the sctri.query will be used.
viewer_heterogeneity – boolean, whether to build the viewer to show the heterogeneity based on one reference annotation. Default: False
viewer_heterogeneity_keys – list, the annotations we want to serve as the reference. Default: None, means the first annotation in sctri.query will be used as the reference.
Examples:
sctri.lazy_run(viewer_heterogeneity_keys=['annotation1','annotation2'])
modality_contributions()¶
- sctriangulate.main_class.ScTriangulate.modality_contributions(self, mode='marker_genes', key='pruned', tops=20, regex_dict={'adt': '^AB_', 'atac': '^chr\\d{1,2}'})¶
calculate the modality contributions for multi modal analysis, the modality contributions of each modality of each cluster means the number of features from this modality that made into the top {tops} feature list. Multiple columns will be added to obs, they are corresponding to the number of modalities considered
- Parameters
mode – string, either ‘marker_genes’ or ‘exclusive_genes’.
key – string, any valid categorical column in self.adata.obs
tops – int, the top n features to consider for each cluster.
regex_dict – dict, keyed by modality name, value is a raw string representing the regex pattern for parsing each modality features
Examples:
sctri.modality_contributions(mode='marker_genes',key='pruned',tops=20)
penalize_artifact()¶
- sctriangulate.main_class.ScTriangulate.penalize_artifact(self, mode, stamps=None, parallel=True)¶
An optional step after running
compute_metricsstep and before thecompute_shapleystep. Basically, we penalize clusters with certain properties by set all their metrics to zero, which forbid them to win in the following “game” step. These undesirable properties can be versatial, for example, cellcylce gene enrichment. We current support two mode:mode1:
void, users specifiy which cluster they want to penalize viastampsparameter.mode2:
cellcycle, program automatically label clusters whose gsea_hit > 5 and gsea_score > 0.8 as invalid cellcyle enriched clusters. And those clusters will be penalized.
- Parameters
mode – string, either ‘void’ or ‘cellcycle’.
stamps – list, contains cluster names that the users want to penalize.
parallel – boolean, whether to run this in parallel (scatter and gather). Default: True.
Examples:
sctri.penalize_artifact(mode='void',stamps=['sctri_rna_leiden_1@c3','sctri_rna_leiden_2@c5']) sctri.penalize_artifact(mode='cellcyle')
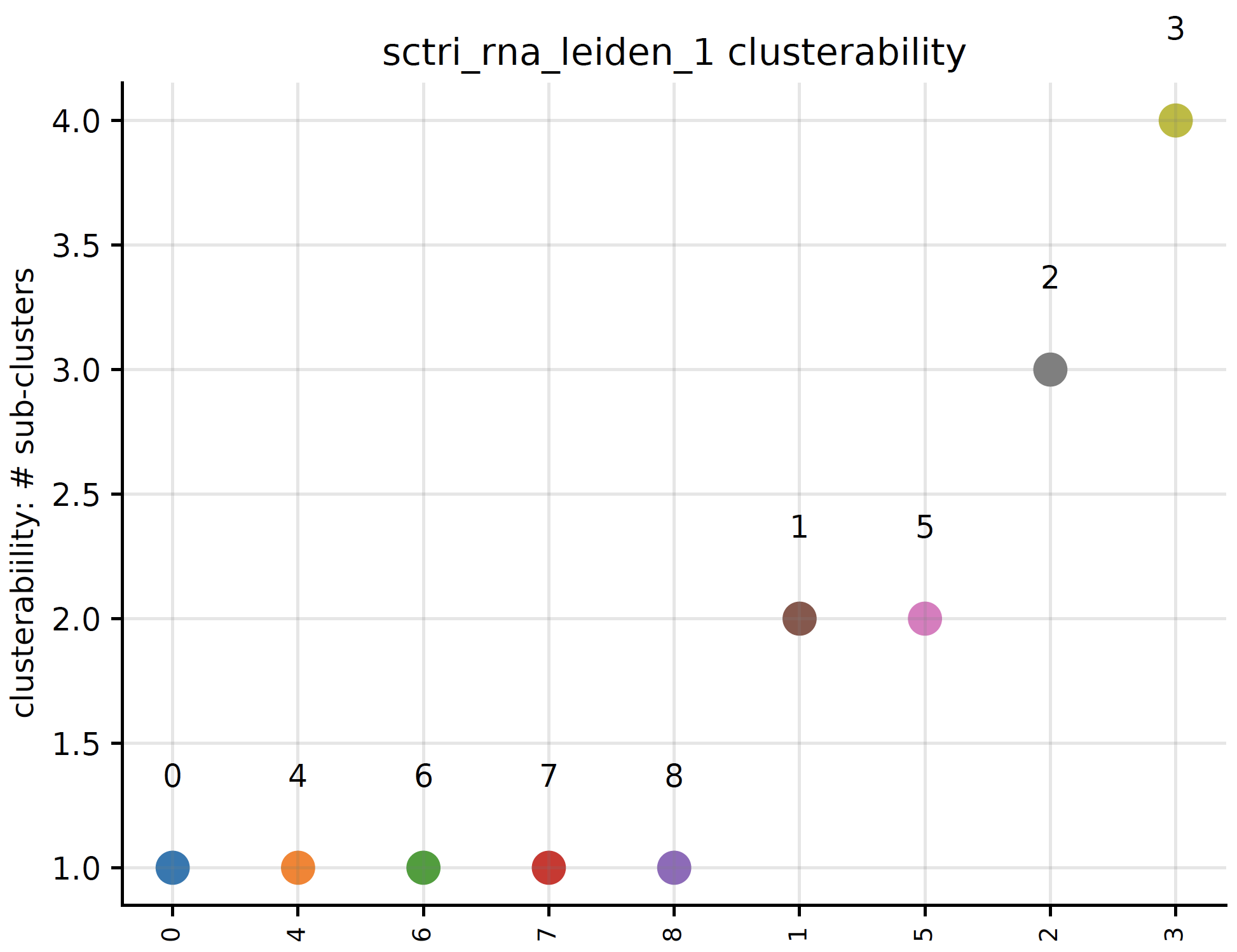
plot_clusterability()¶
- sctriangulate.main_class.ScTriangulate.plot_clusterability(self, key, col, fontsize=3, plot=True, save=True)¶
We define clusterability as the number of sub-clusters the program finds out. If a cluster has being suggested to be divided into three smaller clusters, then the clueterability of this cluster will be 3.
- Parameters
key – string. The clusters from which annotation that you want to assess clusterability.
col – string. Either ‘raw’ cluster or ‘pruned’ cluster.
fontsize – int. The fontsize of x-ticklabels. Default: 3
plot – boolean. Whether to plot the scatterplot or not. Default : True.
save – boolean. Whether to save the plot or not. Default: True
- Returns
python dictionary. {cluster1:#sub-clusters}
Examples:
sctri.plot_clusterability(key='sctri_rna_leiden_1',col='raw',fontsize=8)
plot_cluster_feature()¶
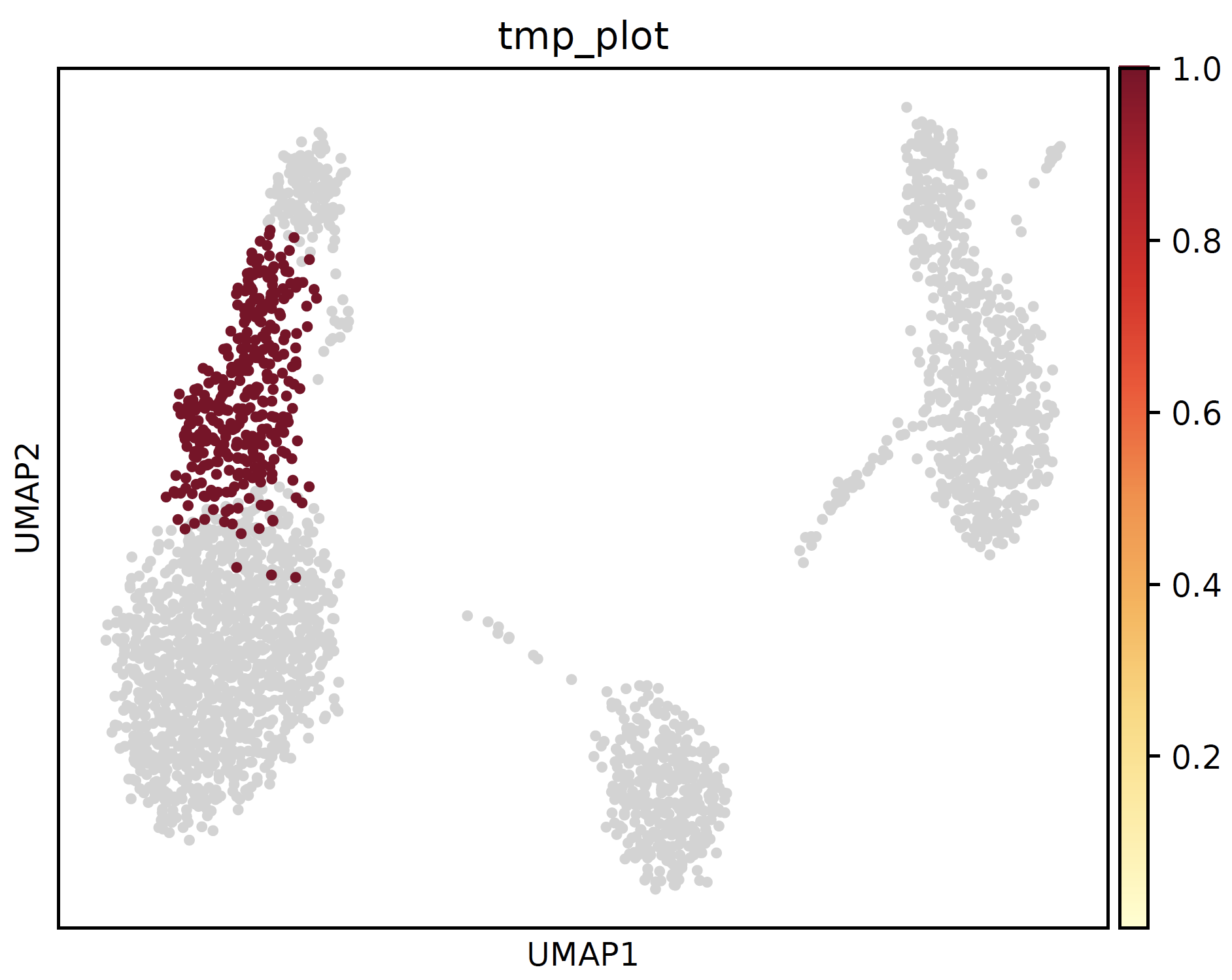
- sctriangulate.main_class.ScTriangulate.plot_cluster_feature(self, key, cluster, feature, enrichment_type='enrichr', save=True, format='pdf')¶
plot the feature of each single clusters, including:
enrichment of artifact genes
marker genes umap
exclusive genes umap
location of clutser umap
- Parameters
key – string. Name of the annation.
cluster – string. Name of the cluster in the annotation.
feature – string, valid value: ‘enrichment’,’marker_genes’, ‘exclusive_genes’, ‘location’
enrichmen_type – string, either ‘enrichr’ or ‘gsea’.
save – boolean, whether to save the figure.
format – string, which format for the saved figure.
Example:
sctri.plot_cluster_feature(key='sctri_rna_leiden_1',cluster='3',feature='enrichment')
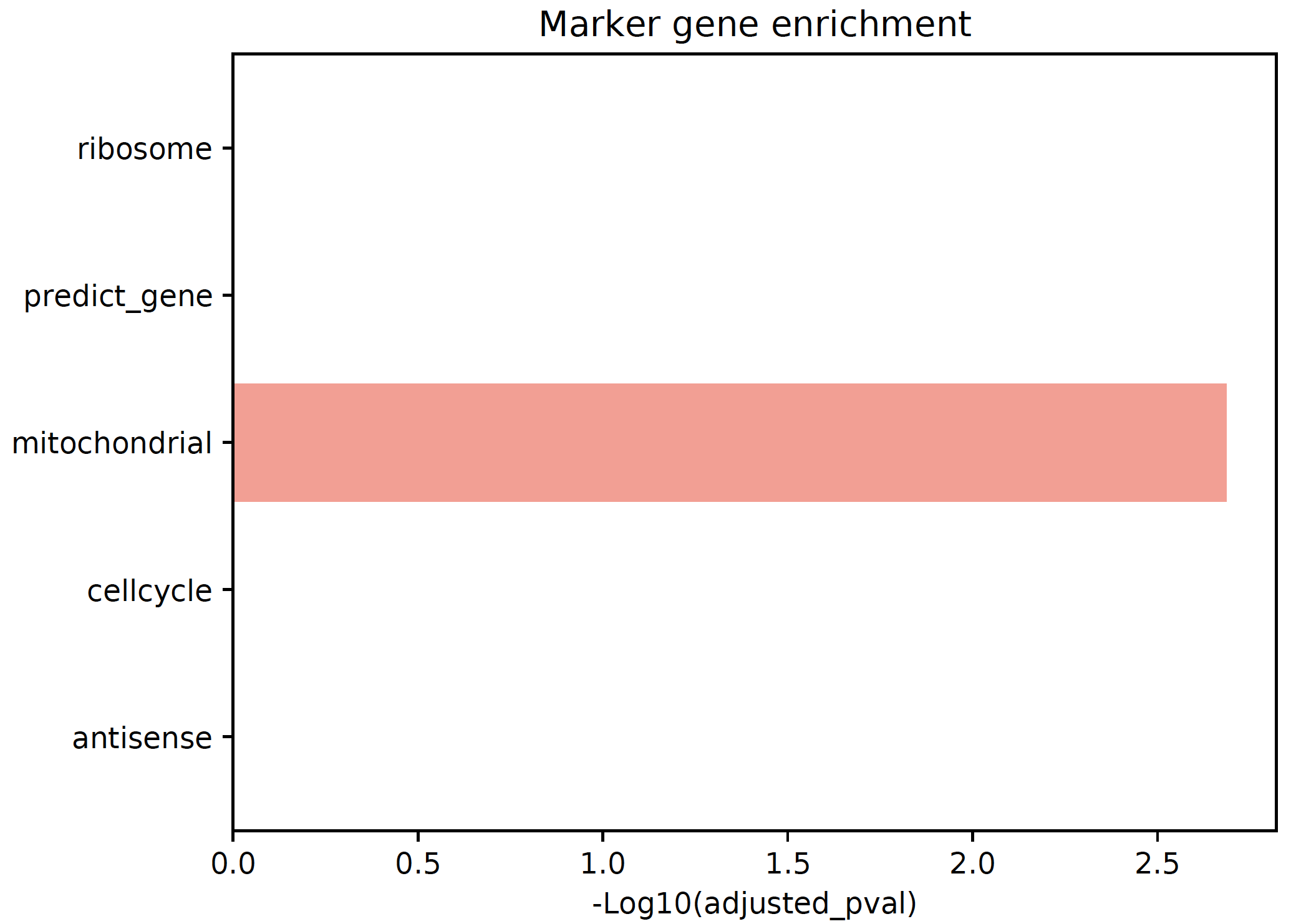
Example:
sctri.plot_cluster_feature(key='sctri_rna_leiden_1',cluster='3',feature='marker_genes')
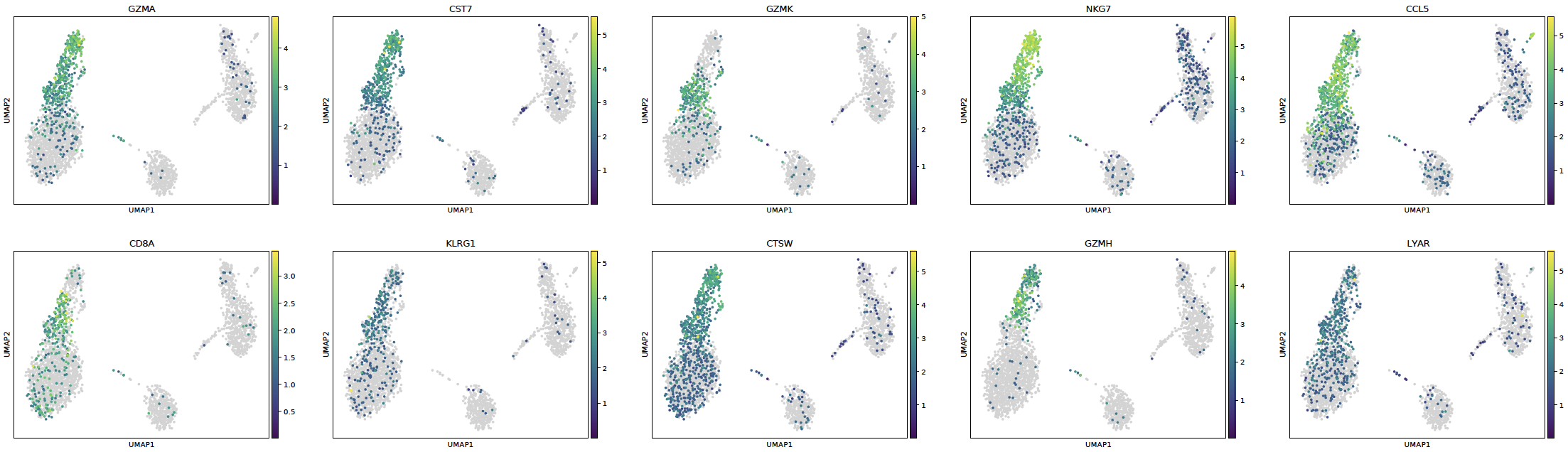
Example:
sctri.plot_cluster_feature(key='sctri_rna_leiden_1',cluster='3',feature='location')
plot_concordance()¶
- sctriangulate.main_class.ScTriangulate.plot_concordance(self, key1, key2, style='3dbar', save=True, format='pdf', cmap=<matplotlib.colors.LinearSegmentedColormap object>, **kwargs)¶
given two annotation, we want to know how the cluster labels from one correponds to the other.
- Parameters
key1 – string, first annotation key
key2 – string, second annotation key
style – string, which style of plot, either ‘heatmap’ or ‘3dbar’
save – boolean, save the figure or not
format – string, format to save
cmap – string or cmap object, the cmap to use for heatmap
- Returns
dataframe, the confusion matrix
Examples:
sctri.plot_concordance(key1='azimuth',key2='pruned',style='3dbar')
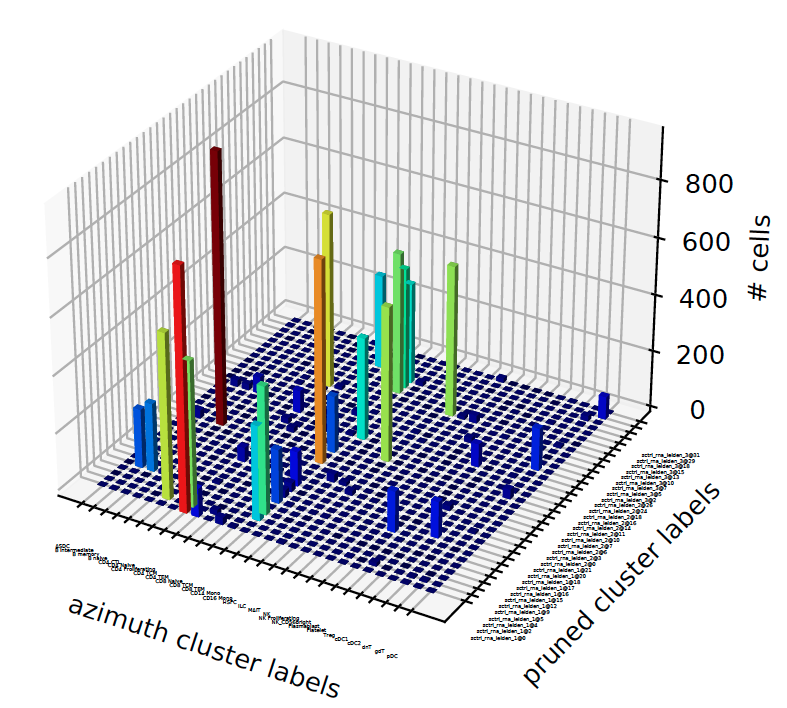
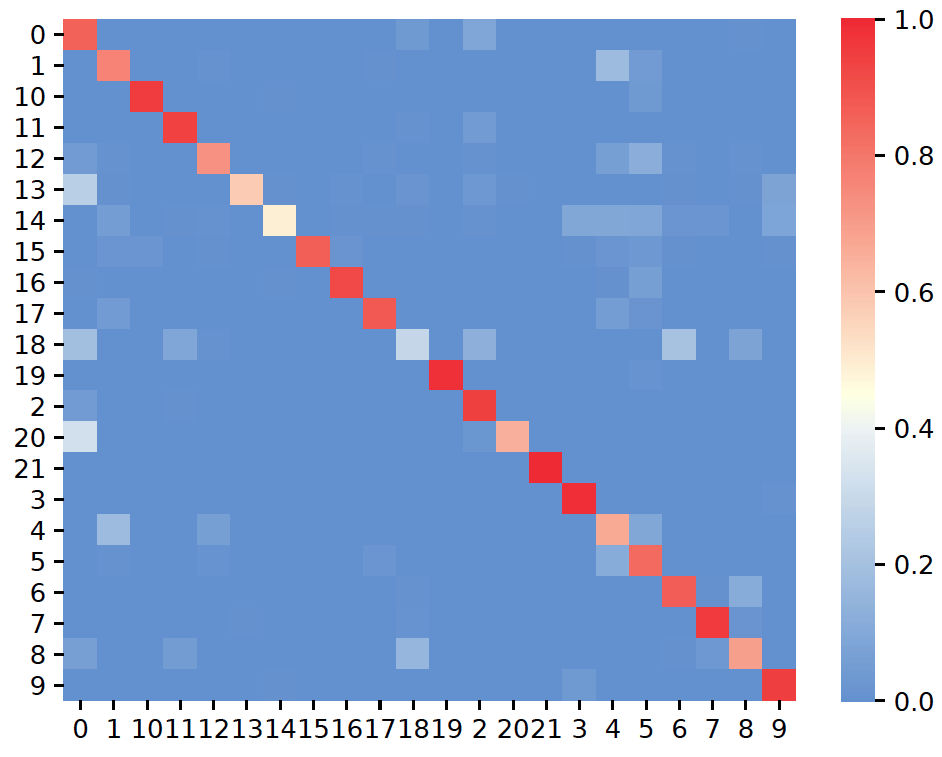
plot_confusion()¶
- sctriangulate.main_class.ScTriangulate.plot_confusion(self, name, key, save=True, format='pdf', cmap=<matplotlib.colors.LinearSegmentedColormap object>, labelsize=None, **kwargs)¶
plot the confusion as a heatmap.
- Parameters
name – string, either ‘confusion_reassign’ or ‘confusion_sccaf’.
key – string, a annotation name which we want to assess the confusion matrix of the clusters.
save – boolean, whether to save the figure. Default: True.
format – boolean, file format to save. Default: ‘.pdf’.
cmap – colormap object, Default: scphere_cmap, which defined in colors module.
labelsize – float, this can adjust the label size on yaxis and xaxis for the resultant heatmap
kwargs – additional keyword arguments to sns.heatmap().
Examples:
sctri.plot_confusion(name='confusion_reassign',key='sctri_rna_leiden_1')
plot_heterogeneity()¶
- sctriangulate.main_class.ScTriangulate.plot_heterogeneity(self, key, cluster, style, col='pruned', save=True, format='pdf', genes=None, umap_zoom_out=True, umap_dot_size=None, subset=None, marker_gene_dict=None, jitter=True, rotation=60, single_gene=None, dual_gene=None, multi_gene=None, merge=None, to_sinto=False, to_samtools=False, cmap='YlOrRd', heatmap_cmap='viridis', heatmap_scale=None, heatmap_regex=None, heatmap_direction='include', heatmap_n_genes=None, heatmap_cbar_scale=None, gene1=None, gene2=None, kind=None, hist2d_bins=50, hist2d_cmap=<matplotlib.colors.ListedColormap object>, hist2d_vmin=1e-05, hist2d_vmax=None, scatter_dot_color='blue', contour_cmap='viridis', contour_levels=None, contour_scatter=True, contour_scatter_dot_size=5, contour_train_kde='valid', surface3d_cmap='coolwarm', **kwarg)¶
Core plotting function in scTriangulate.
- Parameters
key – string, the name of the annotation.
cluster – string, the name of the cluster.
stype –
string, valid values are as below:
umap: plot the umap of this cluster (including its location and its suggestive heterogeneity)
heatmap: plot the heatmap of the differentially expressed features across all sub-populations within this cluster.
build: plot both the umap and heatmap, benefit is the column and raw colorbar of the heatmap is consistent with the umap color
heatmap_custom_gene, plot the heatmap, but with user-defined gene dictionary.
heatmap+umap, it is the umap + heatmap_custom_gene, and the colorbars are matching
violin: plot the violin plot of the specified genes across sub populations.
single_gene: plot the gradient of single genes across the cluster.
dual_gene: plot the dual-gene plot of two genes across the cluster, usually these two genes should correspond to the marker genes in two of the sub-populations.
multi_gene: plot the multi-gene plot of multiple genes across the cluster.
cellxgene: output the h5ad object which are readily transferrable to cellxgene. It also support atac pseudobuld analysis with
to_sintoorto_samtoolsarguments.sankey: plot the sankey plot showing fraction/percentage of cells that flow into each sub population, requiring plotly if you only need html, and kaleido if you need static plot, otherwise, a less pretty matplotlib sankey will be plotted
coexpression: visualize the coexpression pattern of two features, using contour plot or hist2d
col – string, either ‘raw’ or ‘pruned’.
save – boolean, whether to save or not.
foramt – string, which format to save.
genes – list, for violin plot.
umap_zoom_out – boolean, for the umap, whether to zoom out meaning the scale is the same of the whole umap. Zoom in means an amplified version of this cluster.
umap_dot_size – int/float, for the umap.
subset – list, the sub populations we want to keep for plotting.
marker_gene_dict – dict. The custom genes we want the heatmap to display.
jitter – float, for the violin plot.
rotation – int/float, for the violin plot. rotation of the text. Default: 60
single_gene – string, the gene name for single gene plot
dual_gene – list, the dual genes for dual gene plot.
multi_genes – list, the multiple genes for multi gene plot.
merge – nested list, the sub-populations that we want to merge. [(‘sub-c1’,’sub-c2’),(‘sub-c3’,’sub-c4’)]
to_sinto – boolean, for cellxgene mode, output the txt files for running sinto to generate pseudobulk bam files.
to_samtools – boolean,for cellxgene mode, output the txt files for running samtools to generate the pseudobulk bam files.
cmap – a valid string for matplotlib cmap or scTriangulate color module retrieve_pretty_cmap function return object, default is ‘YlOrRd’, will be used for umap
The following will be used for heatmap only:
- Parameters
heatmap_cmap – a valid string for matplotlib cmap or scTriangulate color module retrieve_pretty_cmap function return object, default is ‘viridis’.
heatmap_scale –
None, minmax, median, mean, z_score, default is None, useful when very large or small values exist in the adata.X, scaling can yield better visual effects
Nonemeans no scale will be performed, the raw valus shown in adata.X will be plotted in the heatmapminmaxmeans the raw values will be row-scaled to [0,1] using a MinMaxScalermedianmeans the raw values will be row-scaled via substracting by the median per rowmeanmeans the raw values will be row-scaled via substracting by the mean per rowz_scoremeans the raw values will be row-scaled via Scaling (mean-centered and variance normalized)
heatmap_regex – None or a raw string for example r’^AB_’ (meaning selecing all ADT features as scTriangulate by default prefix ADT features will AB_), the usage of that is to only display certain features from certain modlaities. The underlying implementation is just a regular expression selection.
heatmap_direction – string, ‘include’ or ‘exclude’, it is along with the heatmap_regex parameter, include means doing positive selection, exclude means to exclude the features that match with the heatmap_regex
heatmap_n_genes – an integer, by default, program display 50//n_cluster genes for each cluster, this will overwrite the default.
heatmap_cbar_scale – None or a tuple or a fraction. A tuple for example (-0.5,0.5) will clip the colorbar within -0.5 to 0.5, a fraction number for instance 0.25, will shrink the default colorbar range say -1 to 1 to -0.25 to 0.25
The following will be used for coexpression plot only:
- Parameters
gene1 – the first gene/features to inspect, the gene name, a string.
gene2 – the second gene/features to inspect, the gene name, a string.
kind – a string, ‘scatter’ or ‘hist2d’ or ‘contour’ or ‘contourf’ or ‘surface3d’, those are all supported figure types to represent the coexpression pattern of two features.
hist2d_bins – integer, default is 50, only used is the kind is hist2d, it will determine the number of the bins
hist2d_cmap – a valid matplotlib cmap string, default is bg_greyed_cmap(‘viridis’), only used for hist2d
hist2d_vmin – the min value for hist2d graph, default is 1e-5, useful if you want to make the low expressin region lightgrey.
hist2d_vmax – the max value for hist2d graph, default is None
scatter_dot_color – the color of the scatter plot dot, default is ‘blue’
contour_cmap – the valid matplotlib camp string, default is ‘viridis’
contour_levels – an integer, the levels of contours to show, default is None
contour_scatter – boolean and default is True, whether or not to show the scatter plot on top of the contour plot
contour_scatter_dot_size – float or integer, the dot size of the scatter plot on top of contour plot, default is 5.
contour_train_kde –
a string, either ‘valid’, ‘semi-vaid’ or ‘full’, it determines what subset of dots will be used for inferring the kernel
valid: only data points that are non-zero for both gene1 and gene2semi_valid: only data points that are non-zero for at least one of the genefull: all data points will be used for kde estimation
surface_3d_cmap – a valid matplotlib cmap string, for surface 3d plot, the default would be ‘coolwarm’
Example:
sctri.plot_heterogeneity('leiden1','0','umap',subset=['leiden1@0','leiden3@10']) sctri.plot_heterogeneity('leiden1','0','heatmap',subset=['leiden1@0','leiden3@10']) sctri.plot_heterogeneity('leiden1','0','violin',subset=['leiden1@0','leiden3@10'],genes=['MAPK14','ANXA1']) sctri.plot_heterogeneity('leiden1','0','sankey') sctri.plot_heterogeneity('leiden1','0','cellxgene') sctri.plot_heterogeneity('leiden1','0','heatmap+umap',subset=['leiden1@0','leiden3@10'],marker_gene_dict=marker_gene_dict) sctri.plot_heterogeneity('leiden1','0','dual_gene',dual_gene=['MAPK14','CD52']) sctri.plot_heterogeneity('leiden1','0','coexpression',gene1='MAPK14',genes='CD52',kind='contour')
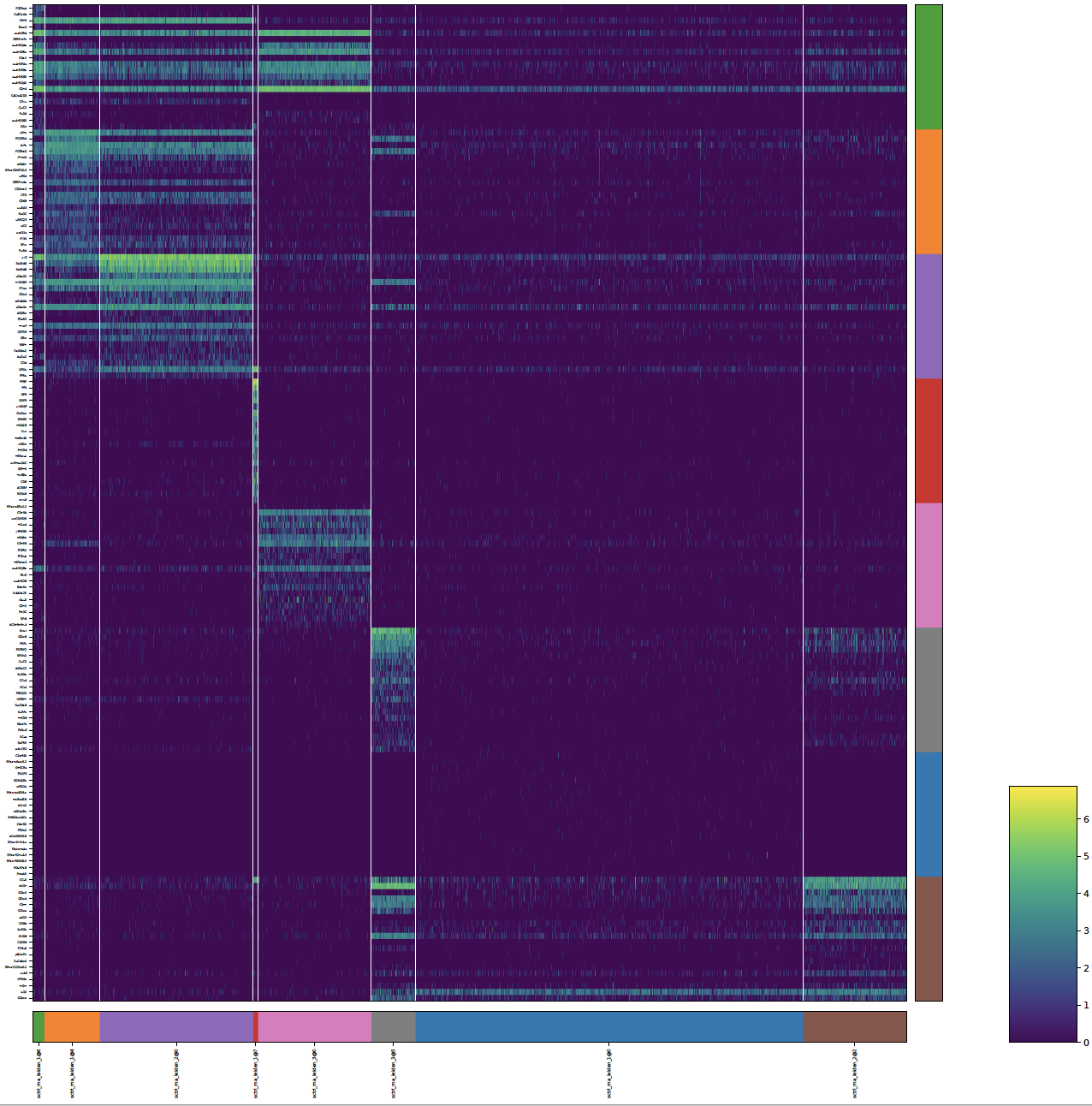
plot_long_heatmap()¶
- sctriangulate.main_class.ScTriangulate.plot_long_heatmap(self, clusters=None, key='pruned', n_features=5, mode='marker_genes', cmap='viridis', save=True, format='pdf', figsize=(6, 4.8), feature_fontsize=3, cluster_fontsize=5, heatmap_regex=None, heatmap_direction='include')¶
the default scanpy heatmap is not able to support the display of arbitrary number of marker genes for each clusters, the max feature is 50. this heatmap allows you to specify as many marker genes for each cluster as possible, and the gene name will all the displayed.
- Parameters
clusters – list, what clusters we want to consider under a certain annotation.
key – string, annotation name.
n_features – int, the number of features to display.
mode – string, either ‘marker_genes’ or ‘exclusive_genes’.
cmap – string, matplotlib cmap string.
save – boolean, whether to save or not.
format – string, which format to save.
figsize – tuple, the width and the height of the plot.
feature_fontsize – int/float. the fontsize for the feature.
cluster_fontsize – int/float, the fontsize for the cluster.
heatmap_regex – None or a raw string for example r’^AB_’ (meaning selecing all ADT features as scTriangulate by default prefix ADT features will AB_), the usage of that is to only display certain features from certain modlaities. The underlying implementation is just a regular expression selection.
heatmap_direction – string, ‘include’ or ‘exclude’, it is along with the heatmap_regex parameter, include means doing positive selection, exclude means to exclude the features that match with the heatmap_regex
Examples:
sctri.plot_long_umap(n_features=20,figsize=(20,20))
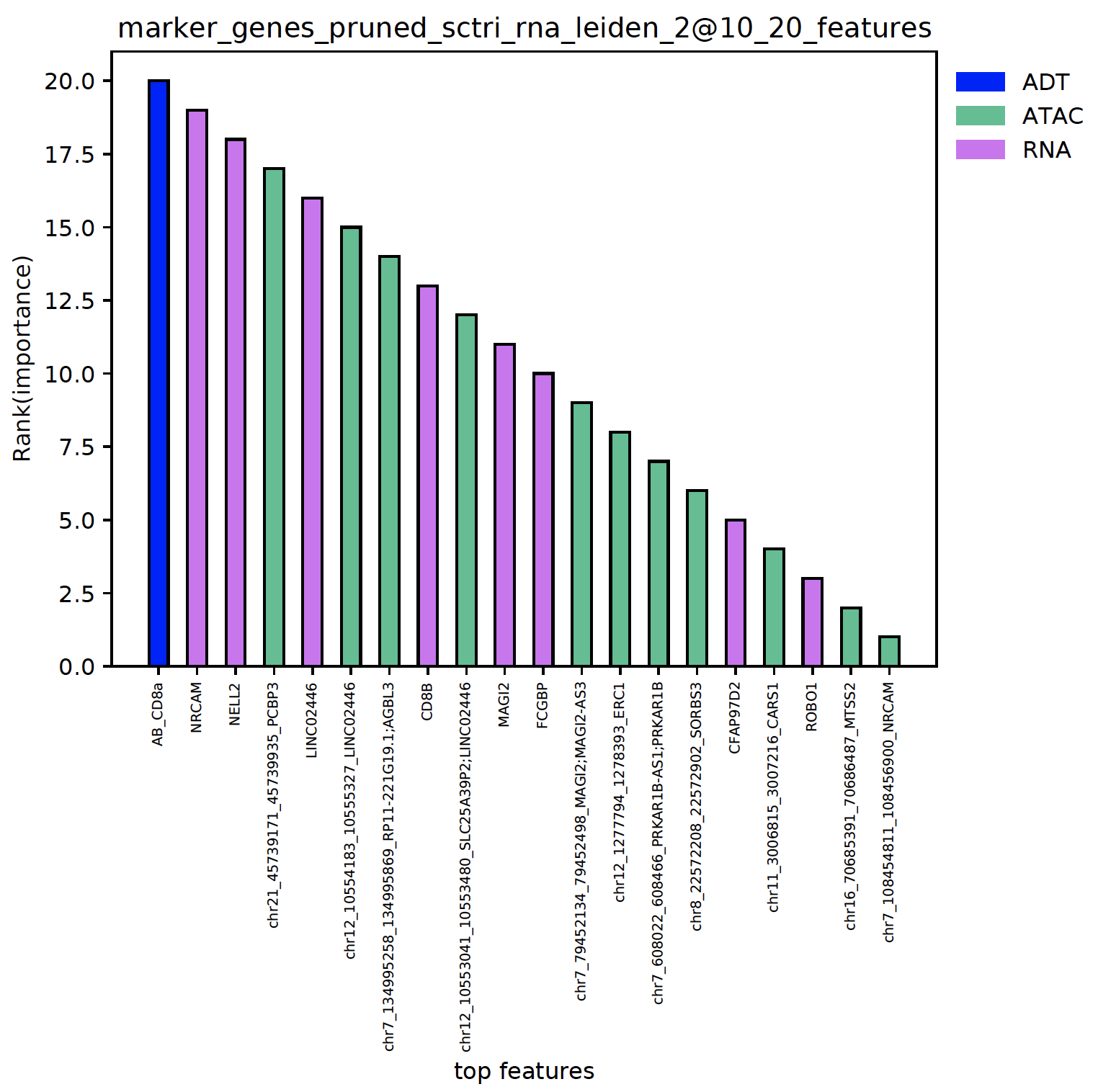
plot_multi_modal_feature_rank()¶
- sctriangulate.main_class.ScTriangulate.plot_multi_modal_feature_rank(self, cluster, mode='marker_genes', key='pruned', tops=20, regex_dict={'adt': '^AB_', 'atac': '^chr\\d{1,2}'}, save=True, format='.pdf')¶
plot the top features in each clusters, the features are colored by the modality and ranked by the importance.
- Parameters
cluster – string, the name of the cluster.
mode – string, either ‘marker_genes’ or ‘exclusive_genes’
tops – int, top n features to plot.
regex_adt – raw string, the pattern by which the ADT feature will be defined.
regex_atac – raw string ,the pattern by which the atac feature will be defined.
save – boolean, whether to save the figures.
format – string, the format the figure will be saved.
Examples:
sctri.plot_multi_modal_feature_rank(cluster='sctri_rna_leiden_2@10')
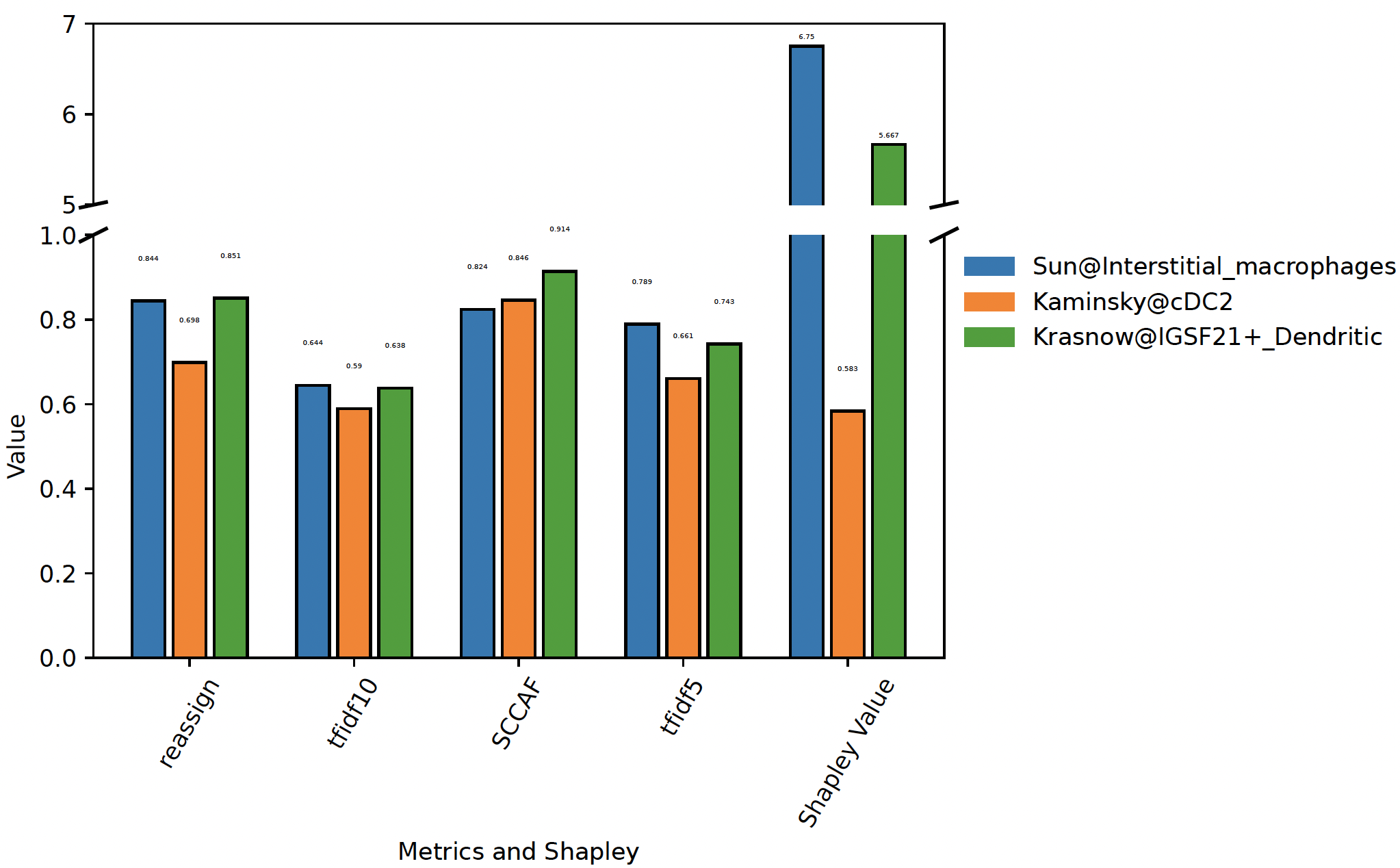
plot_stability()¶
- sctriangulate.main_class.ScTriangulate.plot_stability(self, clusters, broke=True, height_ratios=(0.3, 0.7), hspace=0.1, text_above=0.1, top_ylim=(6, 7), bottom_ylim=(0, 1), break_point_length=0.015)¶
When specifying a list of clutsers, we will plot the stability metrics and shapley values associated with these clustes, This can give an intuitive view regarding which cluster is better
- Parameters
clusters – a list of clusters, each cluster should be annotation@cluster_name
broke – bool, whether to draw barplot with break point or not, default is True
height_ratios – tuple, the height ratios for top ax and bottom ax
hspace – float, the space between top ax and bottom ax
text_above – float, the distance above the bar to draw text
top_ylim – tuple, the ylim of top ax
bottom_ylim – tuple, the ylim of bottom ax
break_point_length – float, to draw a tick to show break point, the length is default to 0.015
Examples:
sctri.plot_stability(clusters=['Sun@Interstitial_macrophages','Kaminsky@cDC2','Krasnow@IGSF21+_Dendritic'],broke=True,top_ylim=[5,7]) sctri.plot_stability(clusters=['Sun@monocytes','Kaminsky@cMonocyte','Kaminsky@ncMonocyte'])
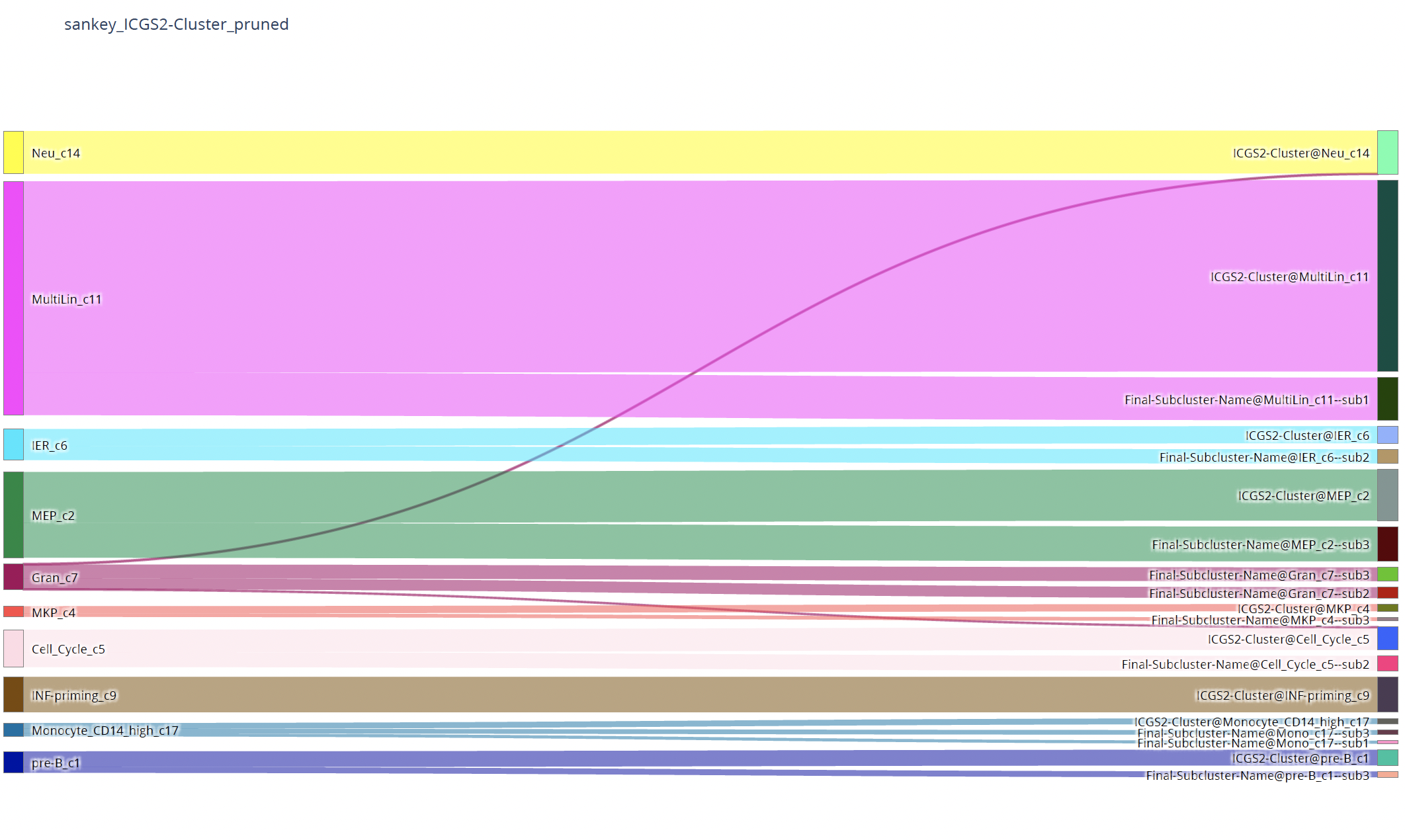
plot_two_column_sankey()¶
- sctriangulate.main_class.ScTriangulate.plot_two_column_sankey(self, left_annotation, right_annotation, opacity=0.6, pad=3, thickness=10, margin=300, text=True, save=True)¶
sankey plot to show the correpondance between two annotation, for example, annotation1 and annotation2, how many cells from each cluster in annotation1 will flow to each cluster in annotation2.
- Parameters
left_annotation – a string, the name of the annotation1
right_annotation – a string, the name of the annotation2
opacity – float number, default is 0.6, the opacity of the sankey strips
pad – float number, default is 3, the gap between blocks vertically
thickness – float number, default is 10, the width of each block
margin – the white margin of the sankey plot, large value means the sankey plot will not consume the whole horizontal space (shrinkaged), default is 300
text – whether to show the text or not, default is True, only set to False if you want to have publication quality static figure, because plotly will add a weired background shady effect on the text, not good for publication, so you can fisrt remove text, then add it back youself manually
save – wheter to save or not, default is True.
Example:
sctri.plot_two_column_sankey('leiden1','leiden2',margin=5)
plot_umap()¶
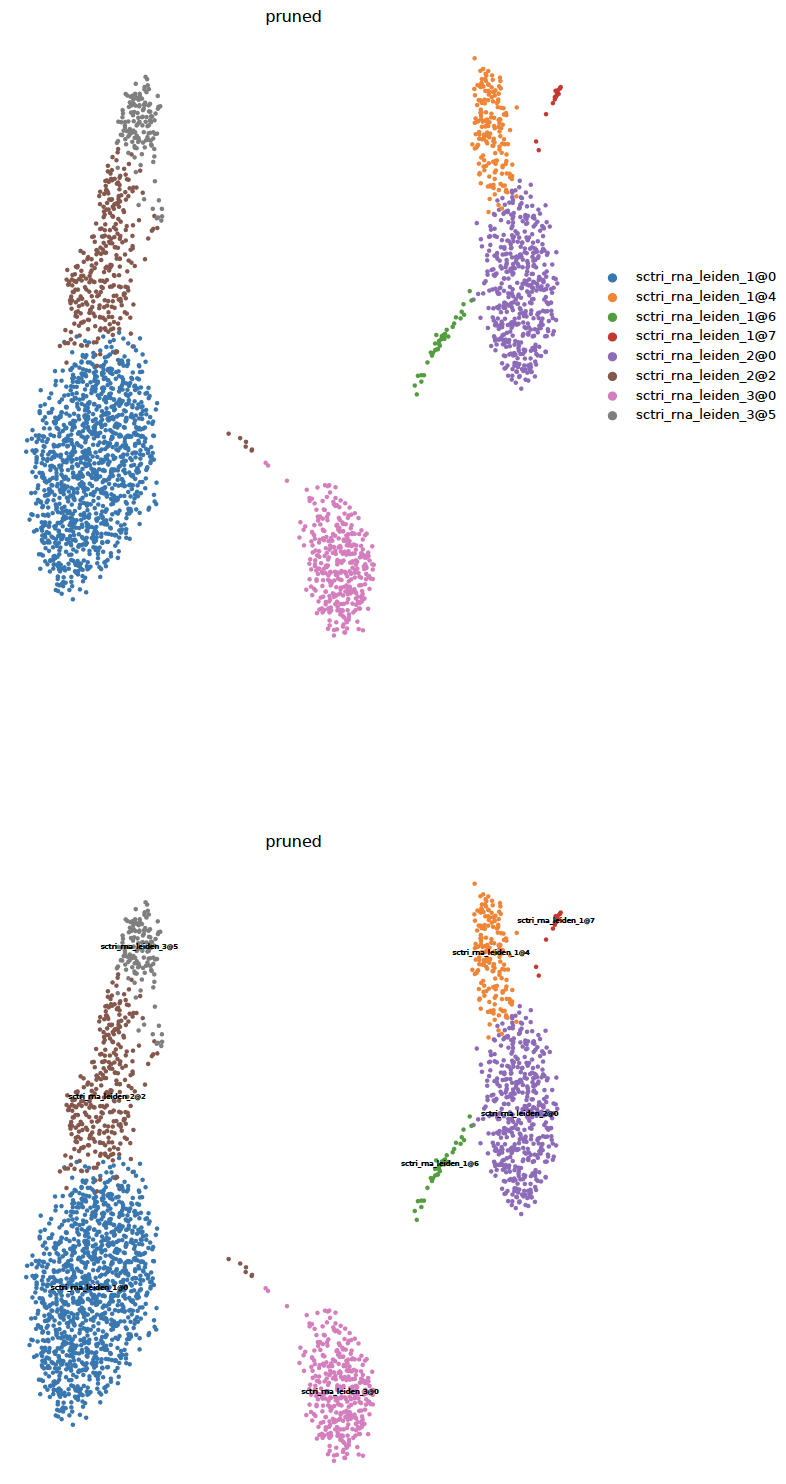
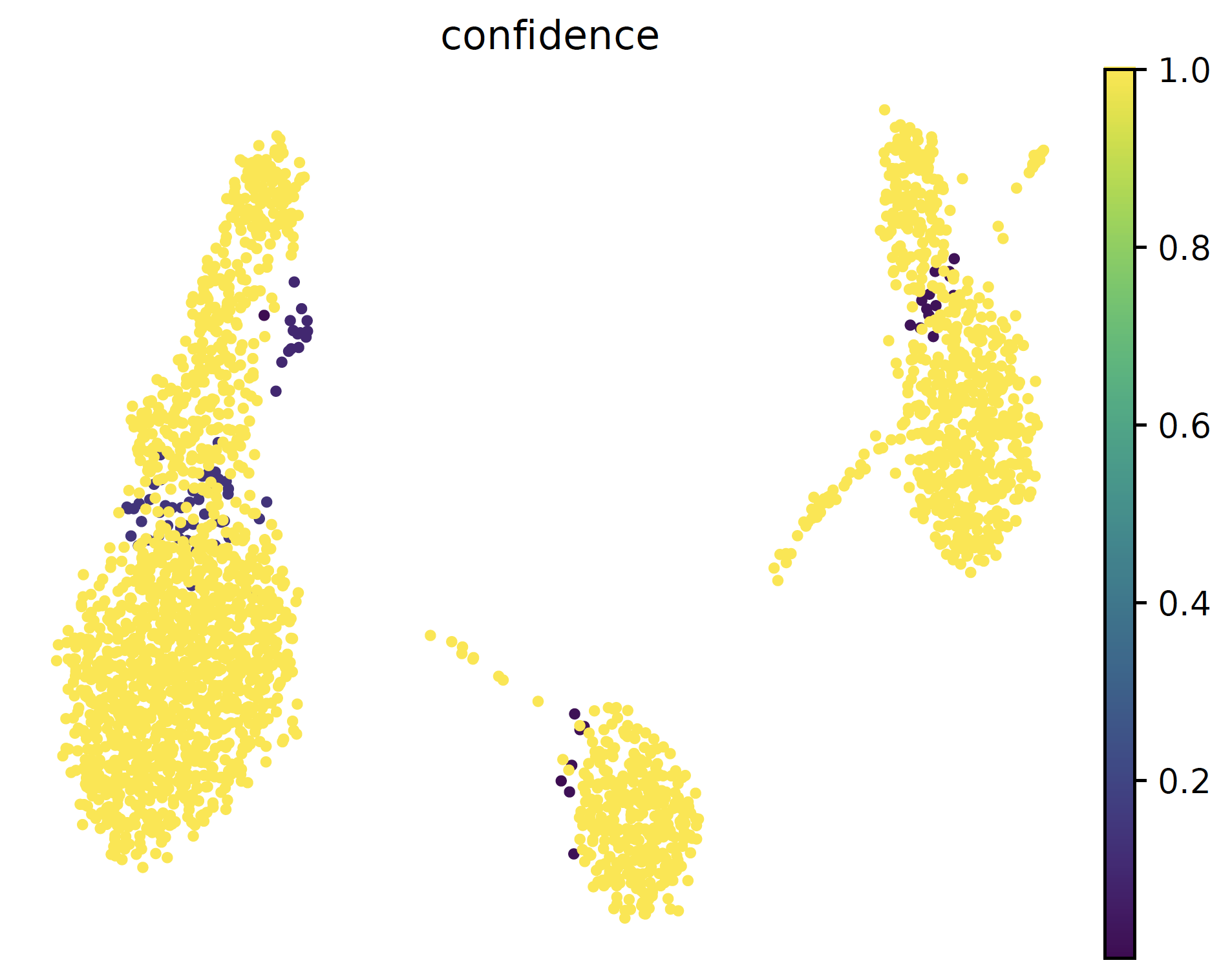
- sctriangulate.main_class.ScTriangulate.plot_umap(self, col, kind='category', save=True, format='pdf', umap_dot_size=None, umap_cmap='YlOrRd', frameon=False)¶
plotting the umap with either category cluster label or continous metrics are important. Different from the scanpy vanilla plot function, this function automatically generate two umap, one with legend on side and another with legend on data, which usually will be very helpful imagine you have > 40 clusters. Secondly, we automatically make all the background dot as light grey, instead of dark color.
- Parameters
col – string, which column in self.adata.obs that we want to plot umap from.
kind – string, either ‘category’ or ‘continuous’
save – boolean, whether to save it to disk or not. Default: True
format – string. Which format to save. Default: ‘.pdf’
umap_dot_size – int/float. the size of dot in scatter plot, if None, using scanpy formula, 120000/n_cells
umap_cmap – string, the matplotlib colormap to use. Default: ‘YlOrRd’
frameon – boolean, whether to have the frame on the umap. Default: False
Examples:
sctri.plot_umap(col='pruned',kind='category') sctri.plot_umap(col='confidence',kind='continous')
plot_winners_statistics()¶
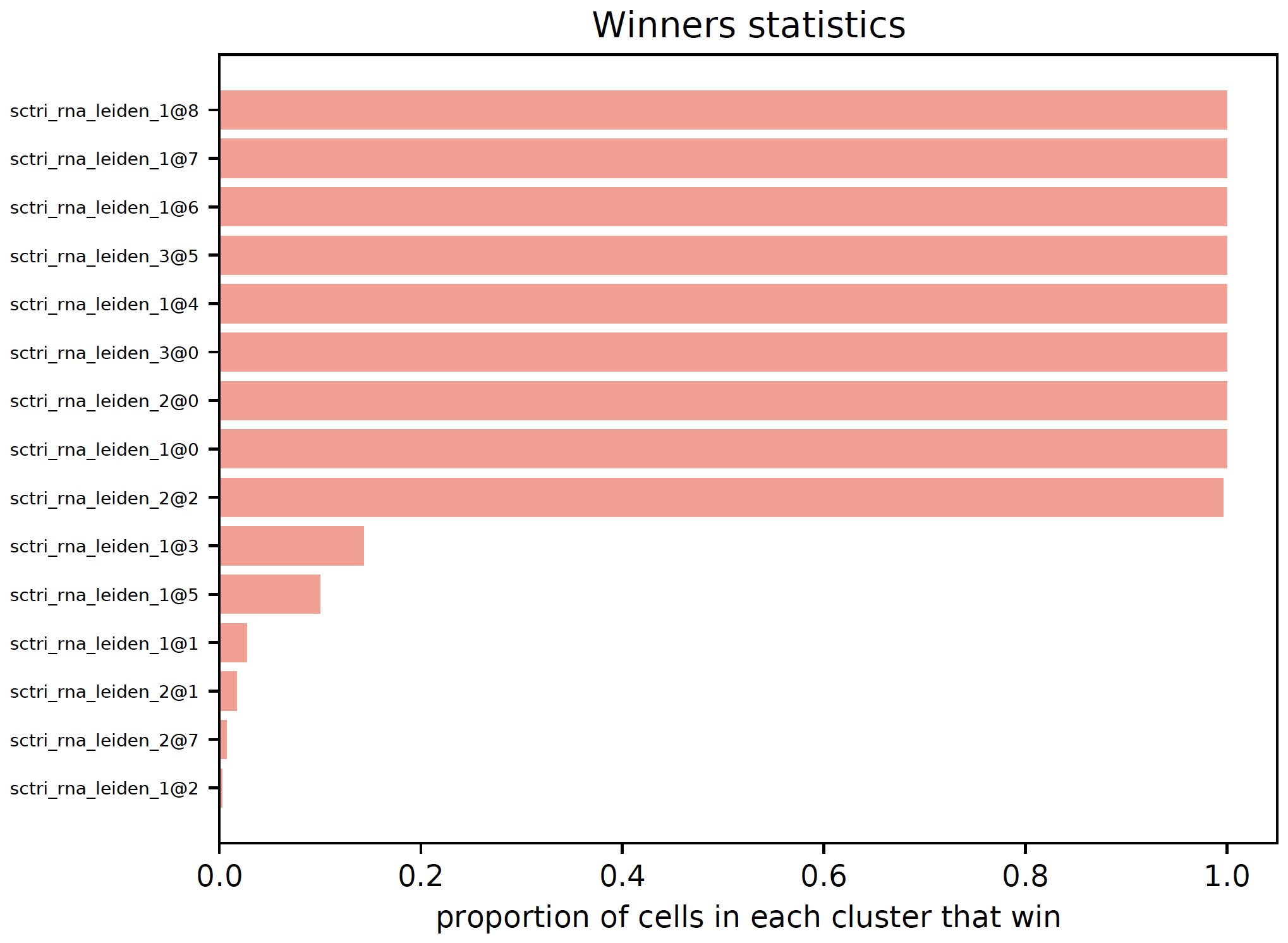
- sctriangulate.main_class.ScTriangulate.plot_winners_statistics(self, col, fontsize=3, plot=True, save=True)¶
For triangulated clusters, either ‘raw’ or ‘pruned’, visualize what fraction of cells won the game. A horizontal barplot will be generated and a dataframe with winners statistics will be returned.
- Parameters
col – string, either ‘raw’ or ‘pruned’
fontsize – int, the fontsize for the y-label. Default: 3
plot – boolean, whether to plot or not. Default: True
save – boolean, whether to save the plot to the sctri.dir or not. Default: True
- Returns
DataFarme
Examples:
sctri.plot_winners_statistics(col='raw',fontsize=4)
pruning()¶
- sctriangulate.main_class.ScTriangulate.pruning(self, method='reassign', discard=None, scale_sccaf=True, layer=None, abs_thresh=10, remove1=True, reference=None, parallel=True, assess_raw=False)¶
Main function. After running
compute_shapley, we get raw cluster results. Althought the raw cluster is informative, there maybe some weired clusters that accidentally win out which doesn’t attribute to its biological stability. For example, a cluster that only has 3 cells, or very unstable cluster. To ensure the best results, we apply a post-hoc assessment onto the raw cluster result, by applying the same set of metrics function to assess the robustness/stability of the raw clusters itself. And we will based on that to perform some pruning to get rid of unstable clusters. Finally, the cells within these clusters will be reassigned to thier nearest neighbors.- Parameters
method – string, valid value: ‘reassign’, ‘rank’.
rankwill compute the metrics on all the raw clusters, together with thediscardparameter which automatically discard clusters ranked at the bottom to remove unstable clusters.reassignwill just remove clusters that either has less thanabs_threshcells or are in the self.invalid attribute list.discard – int. Least {discard} stable clusters to remove. Default: None, means just rank without removing.
scale_sccaf – boolean. whether to scale the expression data. See
compute_metricsfor full explanation. Default: Trueabs_thresh – int. clusters have less than {abs_thresh} cells will be discarded in
reassignmode.remove1 – boolean. When reassign the cells in the dicarded clutsers, whether to also reassign the cells who are the only one in each
referencecluster. Default: Truereference – string. which annotation will serve as the reference.
parallel – boolean, whether to perform this step in parallel. (scatter and gather).Default is True
assess_raw – boolean, whether to run the same set of cluster stability metrics on the raw cluster. Default is False
Examples:
sctri.pruning(method='pruning',discard=None) # just assess and rank the raw clusters sctri.pruning(method='reassign',abs_thresh=10,remove1=True,reference='annotation1') # remove invalid clusters and reassign the cells within
regress_out_size_effect()¶
- sctriangulate.main_class.ScTriangulate.regress_out_size_effect(self, regressor='background_zscore')¶
An optional step to regress out potential confounding effect of cluster_size on the metrics. Run after
compute_metricsstep but beforecompute_shapleystep. All the metrics in selfadata.obs and self.score will be modified in place.- Parameters
regressor – string. which regressor to choose, valid values: ‘background_zscore’, ‘background_mean’, ‘GLM’, ‘Huber’, ‘RANSAC’, ‘TheilSen’
Example:
sctri.regress_out_size(regressor='Huber')
run_single_key_assessment()¶
- sctriangulate.main_class.ScTriangulate.run_single_key_assessment(self, key, scale_sccaf, layer, added_metrics_kwargs)¶
this is a very handy function, given a set of annotation, this function allows you to assess the biogical robustness based on the metrics we define. The obtained score and cluster information will be automatically saved to self.cluster and self.score, and can be further rendered by the scTriangulate viewer.
- Parameters
key – string, the annotation/column name to assess the robustness.
scale_sccaf – boolean, whether to scale the expression data before running SCCAF score. See
compute_metricsfunction for full information.layer – see lazy_run for detail
added_metrics_kwargs – see lazy_run for detail
Examples:
sctri.run_single_key_assessment(key='azimuth',scale_sccaf=True)
serialize()¶
- sctriangulate.main_class.ScTriangulate.serialize(self, name='sctri_pickle.p')¶
serialize the sctri object through pickle protocol to the disk
- Parameters
name – string, the name of the pickle file on the disk. Default: sctri_pickle.p
Examples:
sctri.serialize()
viewer_cluster_feature_figure()¶
- sctriangulate.main_class.ScTriangulate.viewer_cluster_feature_figure(self, parallel=False, select_keys=None, other_umap=None)¶
Generate all the figures for setting up the viewer cluster page.
- Parameters
parallel – boolean, whether to run it in parallel, only work in some linux system, so recommend to not set to True.
select_keys – list, what annotations’ cluster we want to inspect.
other_umap – ndarray,replace the umap with another set.
Examples:
sctri.viewer_cluster_feature_figure(parallel=False,select_keys=['annotation1','annotation2'],other_umap=None)
viewer_cluster_feature_html()¶
- sctriangulate.main_class.ScTriangulate.viewer_cluster_feature_html(self)¶
Setting up the viewer cluster page.
Examples:
sctri.viewer_cluster_feature_html()
viewer_heterogeneity_figure()¶
- sctriangulate.main_class.ScTriangulate.viewer_heterogeneity_figure(self, key, other_umap=None, format='png', heatmap_scale=False, heatmap_cmap='viridis', heatmap_regex=None, heatmap_direction='include', heatmap_n_genes=None, heatmap_cbar_scale=None)¶
Generating the figures for the viewer heterogeneity page
- Parameters
key – string, which annotation to inspect the heterogeneity.
other_umap – ndarray, replace with other umap embedding.
Examples:
sctri.viewer_heterogeneity_figure(key='annotation1',other_umap=None)
Preprocessing Module¶
GeneConvert Class¶
Normalization Class¶
- class sctriangulate.preprocessing.Normalization[source]¶
a series of Normalization functions
Now support:
CLR normalization
total count normalization (CPTT, CPM)
GMM normalization
- static CLR_normalization(mat)[source]¶
Examples:
from sctriangulate.preprocessing import Normalization post_mat = Normalization.CLR_normalization(pre_mat)
- static GMM_normalization(mat, non_negative=False)[source]¶
This method is a re-implementaion from Stephenson et al, The raw counts are first subjected to a CPTT total normalization, then a GaussianMixture model was fitted to the data, we substract the mean of the background from the data to remove background noise. Optionally, user can make the post-processed matrix as non-negative by setting
non_negative==TrueExamples:
from sctriangulate.preprocessing import Normalization post_mat = Normalization.GMM_normalization(pre_mat)
small_txt_to_adata()¶
- sctriangulate.preprocessing.small_txt_to_adata(int_file, gene_is_index=True, sep='\t')[source]¶
given a small dense expression (<2GB) txt file, load them into memory as adata, and also make sure the X is sparse matrix.
- Parameters
int_file – string, path to the input txt file, delimited by tab
gene_is_index – boolean, whether the gene/features are the index
sep – the delimieter, default is tab
- Returns
AnnData
Exmaples:
from sctriangulate.preprocessing import small_txt_to_adata adata= = small_txt_to_adata('./input.txt',gene_is_index=True)
large_txt_to_mtx()¶
- sctriangulate.preprocessing.large_txt_to_mtx(int_file, out_folder, gene_is_index=True, type_convert_to='int16', n_lines=None, sep='\t')[source]¶
Given a large txt dense expression file, convert them to mtx file on cluster to facilitate future I/O
- Parameters
int_file – string, path to the intput txt file, delimited by tab
out_folder – string, path to the output folder where the mtx file will be stored
gene_is_index – boolean, whether the gene/features is the index in the int_file.
type_convert_to – string, since it is a large dataframe, need to read in chunk, to accelarate it and reduce the memory footprint, we convert it to either ‘int16’ if original data is count, or ‘float32’ if original data is normalized data.
n_lines – int, the number of lines for the
int_file, it is just for informative progress barsep – string, defalut is tab, can be other as well
Examples:
from sctriangulate.preprocessing import large_txt_to_mtx large_txt_to_mtx(int_file='input.txt',out_folder='./data',gene_is_index=False,type_convert_to='float32')
mtx_to_adata()¶
- sctriangulate.preprocessing.mtx_to_adata(int_folder, gene_is_index=True, feature='genes.tsv', feature_col='index', barcode='barcodes.tsv', barcode_col='index', matrix='matrix.mtx')[source]¶
convert mtx file to adata in RAM, make sure the X is sparse. Gzip file is supported as well, program will automatically detect gz extension and use gzip pacakge to read it into memory
- Parameters
int_folder – string, folder where the mtx files are stored.
gene_is_index – boolean, whether the gene is index.
feature – string, the name of the feature file, default for rna is genes.tsv
feature_col – ‘index’ as index, or a int (which column, python is zero based) to use in your feature.tsv as feature
barcode – string, the name of the barcode file, default is barcodes.tsv
barcode_col – ‘index’ as index, or a int (which column, python is zero based) to use in your barcodes.tsv as barcode
matrix – string, the name of the sparse matrix
- Returns
AnnData
Examples:
from sctriangulate.preprocessing import mtx_to_adata mtx_to_adata(int_folder='./data',gene_is_index=False,feature='genes')
mtx_to_large_txt()¶
- sctriangulate.preprocessing.mtx_to_large_txt(int_folder, out_file, gene_is_index=False)[source]¶
convert mtx back to large dense txt expression dataframe.
- Parameters
int_folder – string, path to the input mtx folder.
out_file – string, path to the output txt file.
gene_is_index – boolean, whether the gene is the index.
Examples:
from sctriangulate.preprocessing import mtx_to_large_txt mtx_to_large_txt(int_folder='./data',out_file='input.txt',gene_is_index=False)
adata_to_mtx()¶
- sctriangulate.preprocessing.adata_to_mtx(adata, gene_is_index=True, var_column=None, obs_column=None, outdir='data')[source]¶
write a adata to mtx file
- Parameters
adata – AnnData, the adata to convert
gene_is_index – boolean, for the resultant mtx, will gene be the index or feature is the index
var_column – list, the var columns to write the genes.tsv, None will write all available columns
obs_column – list, the obs columns to write the barcodes.tsv, None will write all available columns
outdir – string, the name of the mtx folder, default is data
Example:
adata_to_mtx(adata,True,None,None,'data')
add_azimuth()¶
- sctriangulate.preprocessing.add_azimuth(adata, result, name='predicted.celltype.l2')[source]¶
a convenient function if you have azimuth predicted labels in hand, and want to add the label to the adata.
- Parameters
adata – AnnData
result – string, the path to the ‘azimuth_predict.tsv’ file
name – string, the column name where the user want to transfer to the adata.
Examples:
from sctriangulate.preprocessing import add_azimuth add_azimuth(adata,result='./azimuth_predict.tsv',name='predicted.celltype.l2')
add_annotations()¶
- sctriangulate.preprocessing.add_annotations(adata, inputs, cols_input, index_col=0, cols_output=None, sep='\t', kind='disk')[source]¶
Adding annotations from external sources to the adata
- Parameters
adata – Anndata
inputs – string, path to the txt file where the barcode to cluster label information is stored.
cols_input – list, what columns the users want to transfer to the adata.
index_col – int, for the input, which column will serve as the index column
cols_output – list, corresponding to the cols_input, how these columns will be named in the adata.obs columns
sep – string, default is tab
kind – a string, either ‘disk’, or ‘memory’, disk means the input is the path to the text file, ‘memory’ means the input is the variable name in the RAM that represents the dataframe
Examples:
from sctriangulate.preprocessing import add_annotations add_annotations(adata,inputs='./annotation.txt',cols_input=['col1','col2'],index_col=0,cols_output=['annotation1','annontation2'],kind='disk') add_annotations(adata,inputs=df,cols_input=['col1','col2'],index_col=0,cols_output=['annotation1','annontation2'],kind='memory')
add_umap()¶
- sctriangulate.preprocessing.add_umap(adata, inputs, mode, cols=None, index_col=0, key='X_umap')[source]¶
if umap embedding is pre-computed, add it back to adata object.
- Parameters
adata – Anndata
inputs – string, path to the the txt file where the umap embedding was stored.
mode –
string, valid value ‘pandas_disk’, ‘pandas_memory’, ‘numpy’
pandas_disk: the inputs argument should be the path to the txt file
pandas_memory: the inputs argument should be the name of the pandas dataframe in the program, inputs=df
numpy, the inputs argument should be a 2D ndarray contains pre-sorted (same order as barcodes in adata) umap coordinates
cols – list, what columns contain umap embeddings
index_col – int, which column will serve as the index column.
key – string, the key name to add to obsm
Examples:
from sctriangulate.preprocessing import add_umap add_umap(adata,inputs='umap.txt',mode='pandas_disk',cols=['umap1','umap2'],index_col=0)
doublet_predict()¶
- sctriangulate.preprocessing.doublet_predict(adata)[source]¶
wrapper function for running srublet, a new column named ‘doublet_scores’ will be added to the adata
- Parameters
adata – Anndata
- Returns
dict
Examples:
from sctriangulate.preprocessing import doublet_predict mapping = doublet_predict(old_adata)
make_sure_adata_writable()¶
- sctriangulate.preprocessing.make_sure_adata_writable(adata, delete=False)[source]¶
maks sure the adata is able to write to disk, since h5 file is stricted typed, so no mixed dtype is allowd. this function basically is to detect the column of obs/var that are of mixed types, and delete them.
- Parameters
adata – Anndata
delete – boolean, False will just print out what columns are mixed type, True will automatically delete those columns
- Returns
Anndata
Examples:
from sctriangulate.preprocessing import make_sure_adata_writable make_sure_adata_writable(adata,delete=True)
just_log_norm()¶
scanpy_recipe()¶
- sctriangulate.preprocessing.scanpy_recipe(adata, species='human', is_log=False, resolutions=[1, 2, 3], modality='rna', umap=True, save=True, pca_n_comps=None, n_top_genes=3000)[source]¶
Main preprocessing function. Run Scanpy normal pipeline to achieve Leiden clustering with various resolutions across multiple modalities.
- Parameters
adata – Anndata
species – string, ‘human’ or ‘mouse’
is_log – boolean, whether the adata.X is count or normalized data.
resolutions – list, what leiden resolutions the users want to obtain.
modality – string, valid values: ‘rna’,’adt’,’atac’, ‘binary’[mutation data, TCR data, etc]
umap – boolean, whether to compute umap embedding.
save – boolean, whether to save the obtained adata object with cluster label information in it.
pca_n_comps – int, how many PCs to keep when running PCA. Suggestion: RNA (30-50), ADT (15), ATAC (100)
n_top_genes – int, how many features to keep when selecting highly_variable_genes. Suggestion: RNA (3000), ADT (ignored), ATAC (50000-100000)
- Returns
Anndata
Examples:
from sctriangulate.preprocessing import scanpy_recipe # rna adata = scanpy_recipe(adata,is_log=False,resolutions=[1,2,3],modality='rna',pca_n_comps=50,n_top_genes=3000) # adt adata = scanpy_recipe(adata,is_log=False,resolutions=[1,2,3],modality='adt',pca_n_comps=15) # atac adata = scanpy_recipe(adata,is_log=False,resolutions=[1,2,3],modality='atac',pca_n_comps=100,n_top_genes=100000) # binary adata = scanpy_recipe(adata,resolutions=[1,2,3],modality='binary')
concat_rna_and_other()¶
- sctriangulate.preprocessing.concat_rna_and_other(adata_rna, adata_other, umap, umap_key, name, prefix)[source]¶
concatenate rna adata and another modality’s adata object
- Parameters
adata_rna – AnnData
adata_other – Anndata
umap – string, whose umap to use, either ‘rna’ or ‘other’
umap_key – string, either ‘X_umap’ or ‘X_tsne’ or other string for obsm key
name – string, the name of other modality, for example, ‘adt’ or ‘atac’
prefix – string, the prefix added in front of features from other modality, by scTriangulate convertion, adt will be ‘AB_’, atac will be ‘’.
- Return adata_combine
Anndata
Examples:
from sctriangulate.preprocessing import concat_rna_and_other concat_rna_and_other(adata_rna,adata_adt,umap='rna',name='adt',prefix='AB_')
nca_embedding()¶
- sctriangulate.preprocessing.nca_embedding(adata, nca_n_components, label, method, max_iter=50, plot=True, save=True, format='pdf', legend_loc='on data', n_top_genes=None, hv_features=None, add_features=None)[source]¶
Doing Neighborhood component ananlysis (NCA), so it is a supervised PCA that takes the label from the annotation, and try to generate a UMAP embedding that perfectly separate the labelled clusters.
- Parameters
adata – the Anndata
nca_n_components – recommend to be 10 based on Ref
label – string, the column name which contains the label information
method – either ‘umap’ or ‘tsne’
max_iter – for the NCA, default is 50, it is generally good enough
plot – whether to plot the umap/tsne or not
save – whether to save the plot or not
format – the saved format, default is ‘pdf’
legend_loc – ‘on data’ or ‘right margin’
n_top_genes – how many hypervariable genes to choose for NCA, recommended 3000 or 5000, default is None, means there will be other features to add, multimodal setting
hv_features – a list contains the user-supplied hypervariable genes/features, in multimodal setting, this can be [rna genes] + [ADT protein]
add_features – this should be another adata contains features from other modalities, or None means just for RNA
Example:
from sctriangulate.preprocessing import nca_embedding # only RNA nca_embedding(adata,nca_n_components=10,label='annotation1',method='umap',n_top_genes=3000) # RNA + ADT # list1 contains [gene features that are variable] and [ADT features that are variable] nca_embedding(adata_rna,nca_n_components=10,label='annotation1',method='umap',n_top_genes=3000,hv_features=list1, add_features=adata_adt)
umap_dual_view_save()¶
- sctriangulate.preprocessing.umap_dual_view_save(adata, cols, method='umap')[source]¶
generate a pdf file with two umap up and down, one is with legend on side, another is with legend on data. More importantly, this allows you to generate multiple columns iteratively.
- Parameters
adata – Anndata
cols – list, all columns from which we want to draw umap.
method – string, either umap or tsne
Examples:
from sctriangulate.preprocessing import umap_dual_view_save umap_dual_view_save(adata,cols=['annotation1','annotation2','total_counts'])
umap_color_exceed_102()¶
- sctriangulate.preprocessing.umap_color_exceed_102(adata, key, dot_size=None, legend_fontsize=6, outdir='.', name=None, **kwargs)[source]¶
draw a umap that bypass the scanpy 102 color upper bound, this can generate as many as 433 clusters.
- Parameters
adata – Anndata
key – the categorical column in adata.obs, which will be plotted
dot_size – None or number
legend_fontsize – defualt is 6
outdir – output directory, default is ‘.’
name – name of the plot, default is None
Exmaple:
from sctriangulate.preprocessing import umap_color_exceed_102 umap_color_exceed_102(adata,key='leiden6') # more than 130 clusters
make_sure_mat_dense()¶
make_sure_mat_sparse()¶
gene_activity_count_matrix_old_10x()¶
- sctriangulate.preprocessing.gene_activity_count_matrix_old_10x(fall_in_promoter, fall_in_gene, valid=None)[source]¶
this function is to generate gene activity count matrix, please refer to
gene_activity_count_matrix_new_10xfor latest version of 10x fragements.tsv output.how to get these two arguments? (LIGER team approach)
sort the fragment, gene and promoter bed, or use function in this module to sort the reference bed files:
sort -k1,1 -k2,2n -k3,3n pbmc_granulocyte_sorted_10k_atac_fragments.tsv > atac_fragments.sort.bed sort -k 1,1 -k2,2n -k3,3n hg19_genes.bed > hg19_genes.sort.bed sort -k 1,1 -k2,2n -k3,3n hg19_promoters.bed > hg19_promoters.sort.bed
bedmap:
module load bedops bedmap --ec --delim " " --echo --echo-map-id hg19_promoters.sort.bed atac_fragments.sort.bed > atac_promoters_bc.bed bedmap --ec --delim " " --echo --echo-map-id hg19_genes.sort.bed atac_fragments.sort.bed > atac_genes_bc.bed
the following was taken from http://htmlpreview.github.io/?https://github.com/welch-lab/liger/blob/master/vignettes/Integrating_scRNA_and_scATAC_data.html
delim. This changes output delimiter from ‘|’ to indicated delimiter between columns, which in our case is “ ”.
ec. Adding this will check all problematic input files.
echo. Adding this will print each line from reference file in output. The reference file in our case is gene or promoter index.
echo-map-id. Adding this will list IDs of all overlapping elements from mapping files, which in our case are cell barcodes from fragment files.
Finally:
from sctriangulate.preprocessing import gene_activity_count_matrix_old_10x gene_activity_count_matrix_old_10x(fall_in_promoter,fall_in_gene,valid=None)
gene_activity_count_matrix_new_10x()¶
- sctriangulate.preprocessing.gene_activity_count_matrix_new_10x(fall_in_promoter, fall_in_gene, valid=None)[source]¶
Full explanation please refer to
gene_activity_count_matrix_old_10xExamples:
from sctriangulate.preprocessing import gene_activity_count_matrix_new_10x gene_activity_count_matrix_new_10x(fall_in_promoter,fall_in_gene,valid=None)
format_find_concat()¶
- sctriangulate.preprocessing.format_find_concat(adata, canonical_chr_only=True, gtf_file='gencode.v38.annotation.gtf', key_added='gene_annotation', **kwargs)[source]¶
this is a wrapper function to add nearest genes to your ATAC peaks or bins. For instance, if the peak is chr1:55555-55566, it will be annotated as chr1:55555-55566_gene1;gene2 :param adata: The anndata, the var_names is the peak/bin, please make sure the format is like chr1:55555-55566 :param canonical_chr_only: boolean, default to True, means only contain features on canonical chromosomes. for human, it is chr1-22 and X,Y :param gtf_file: the path to the gtf files, we provide the hg38 on this google drive link to download :param key_added: string, the column name where the gene annotation will be inserted to adata.var, default is ‘gene_annotation’ :return adata: Anndata, the gene annotation will be added to var, and the var_name will be suffixed with gene annotation, if canonical_chr_only is True, then only features on canonical
chromsome will be retained.
- Example::
adata = format_find_concat(adata)
find_genes()¶
- sctriangulate.preprocessing.find_genes(adata, gtf_file, key_added='gene_annotation', upstream=2000, downstream=0, feature_type='gene', annotation='HAVANA', raw=False)[source]¶
This function is taken from episcanpy. all the credits to the original developer
merge values of peaks/windows/features overlapping genebodies + 2kb upstream. It is possible to extend the search for closest gene to a given number of bases downstream as well. There is commonly 2 set of annotations in a gtf file(HAVANA, ENSEMBL). By default, the function will search annotation from HAVANA but other annotation label/source can be specifed. It is possible to use other type of features than genes present in a gtf file such as transcripts or CDS.
Examples:
from sctriangulate.preprocessing import find_genes find_genes(adata,gtf_file='gencode.v38.annotation.gtf)
reformat_peak()¶
- sctriangulate.preprocessing.reformat_peak(adata, canonical_chr_only=True)[source]¶
To use
find_genesfunction, please first reformat the peak from 10X format “chr1:10109-10357” to find_gene format “chr1_10109_10357”- Parameters
adata – AnnData
canonical_chr_only – boolean, only kept the canonical chromosome
- Returns
AnnData
Examples:
from sctriangulate.preprocessing import reformat_peak adata = reformat_peak(adata,canonical_chr_only=True)
rna_umap_transform()¶
- sctriangulate.preprocessing.rna_umap_transform(outdir, ref_exp, ref_group, q_exp_list, q_group_list, q_identifier_list, pca_n_components, umap_min_dist=0.5)[source]¶
Take a reference expression matrix (pandas dataframe), and a list of query expression matrics (pandas dataframe), along with a list of query expression identifiers. This function will generate the umap transformation of your reference exp, and make sure to squeeze your each query exp to the same umap transformation.
- Parameters
outdir – the path in which all the results will go
ref_exp – the pandas dataframe object,index is the features, column is the cell barcodes
ref_group – the pandas series object, index is the cell barcodes, the value is the clusterlabel information
q_exp_list – the list of pandas dataframe object, requirement is the same as ref_exp
q_group_list – the list of pandas series object, requirement is the same as ref_group
q_identifier_list – the list of string, to denote the name of each query dataset
pca_n_components – the int, denote the PCs to use for PCA
umap_min_dist – the float number, denote the min_dist parameter for umap program
Examples:
rna_umap_transform(outdir='.',ref_exp=ref_df,ref_group=ref_group_df,q_exp_list=[q1_df],q_group_list=[q1_group_df],q_identifier_list=['q1'],pca_n_components=50)
Colors Module¶
color_stdout()¶
- sctriangulate.colors.color_stdout(skk, c)[source]¶
color your output to the terminal :param skk: the string you want to color :param c: the name of the color ‘red’,’green’,’yellow’,’lightpurple’,’cyan’,’lightgrey’,’black’
Example:
from color import color_stdout # when print to terminal, it will be red print(color_stdout('hello','red'))
pick_n_colors()¶
- sctriangulate.colors.pick_n_colors(n, gradient=False, cmap=None)[source]¶
a very handy and abstract function, pick n colors in hex code that guarantee decent contrast.
n <=10, use tab10
10 < n <= 20, use tab20
20 < n <= 28, use zeileis (take from scanpy)
28 < n <= 102, use godsnot (take from scanpy)
n > 102, use jet cmap (no guarantee for obvious contrast)
- Parameters
n – int, how many colors are needed
gradient – boolean, whether to use gradient color, default is False
cmap – string, the valid cmap to use if gradient=True
- Returns
list, each item is a hex code.
Examples:
generate_block(color_list = pick_n_colors(10),name='tab10') generate_block(color_list = pick_n_colors(20),name='tab20') generate_block(color_list = pick_n_colors(28),name='zeileis') generate_block(color_list = pick_n_colors(102),name='godsnot') generate_block(color_list = pick_n_colors(200),name='433')

colors_for_set()¶
- sctriangulate.colors.colors_for_set(setlist, **kwargs)[source]¶
given a set of items, based on how many unique item it has, pick the n color
- Parameters
setlist – list without redundant items.
**kwargs –
will be passed to pick_n_colors function
- Returns
dictionary, {each item: hex code}
Exmaples:
cmap_dict = colors_for_set(['batch1','batch2]) # {'batch1': '#1f77b4', 'batch2': '#ff7f0e'}
gradienting()¶
- sctriangulate.colors.gradienting(input_hex, n)[source]¶
Given a hex color code (pivot color), it returns a gradient (specified by n) determined by this pivot color
- Parameters
input_hex – string, like ‘#4c4cff’
n – int, how many gradient you want
- Return gradiented_hex
list, like [‘#d2d2ff’, ‘#a5a5ff’, ‘#7878ff’, ‘#4c4cff’]
Examples:
gradiented_hex = gradienting('#4c4cff',n=4) # ['#d2d2ff', '#a5a5ff', '#7878ff', '#4c4cff']
bg_greyed_cmap()¶
- sctriangulate.colors.bg_greyed_cmap(cmap_str)[source]¶
set 0 value as lightgrey, which will render better effect on umap
- Parameters
cmap_str – string, any valid matplotlib colormap string
- Returns
colormap object
Examples:
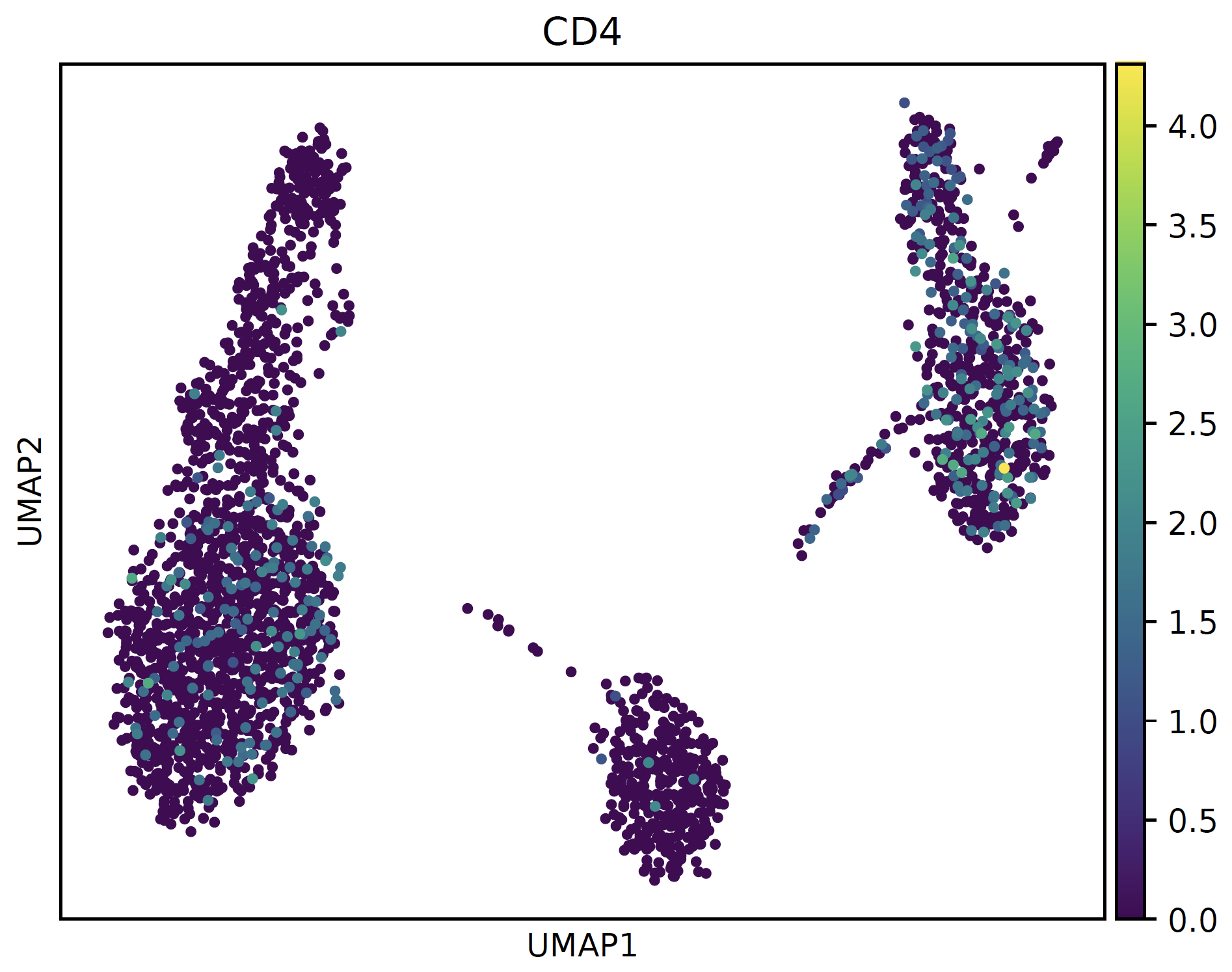
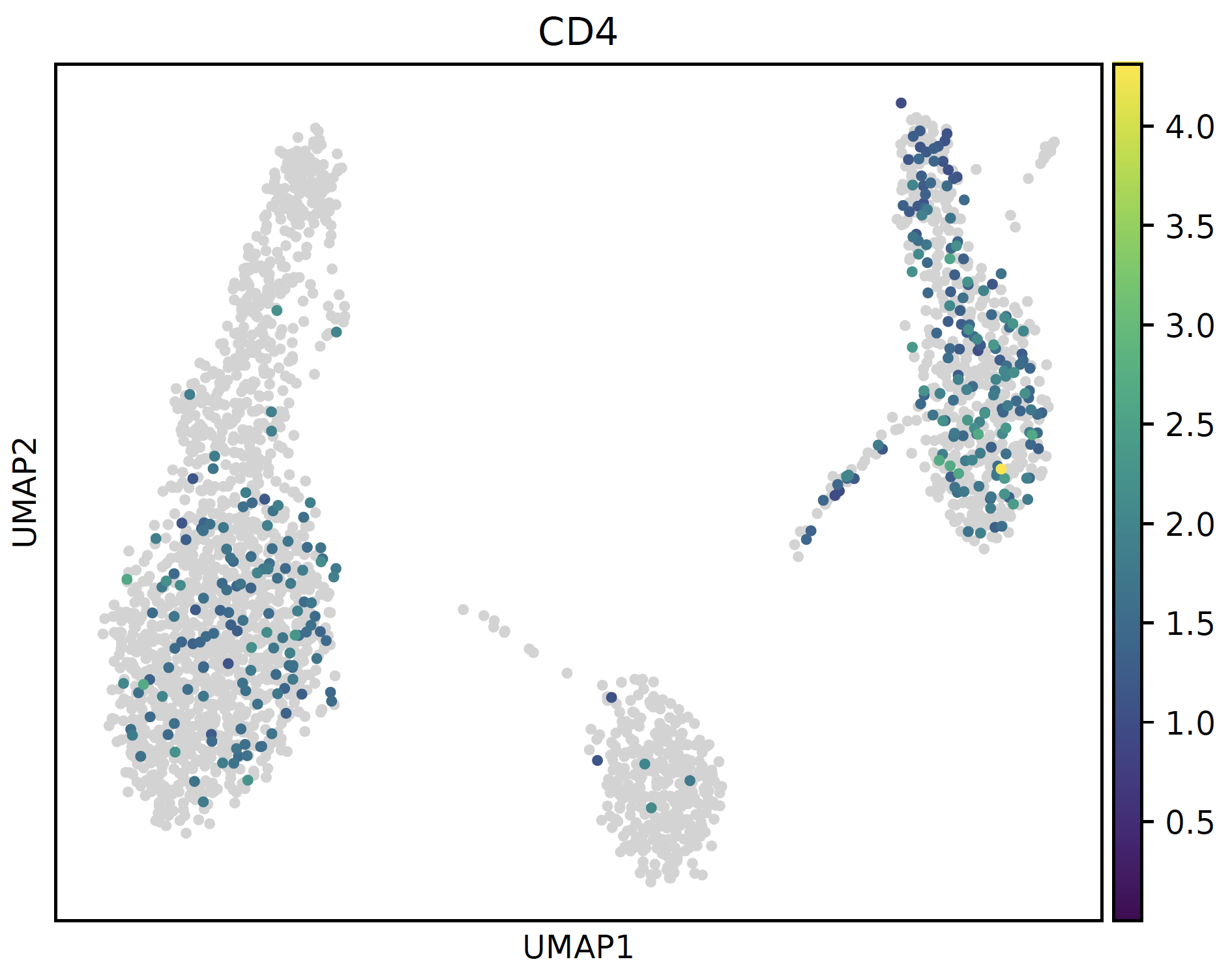
# normal cmap sc.pl.umap(sctri.adata,color='CD4',cmap='viridis') plt.savefig('normal.pdf',bbox_inches='tight') plt.close() # bg_greyed cmap sc.pl.umap(sctri.adata,color='CD4',cmap=bg_greyed_cmap('viridis'),vmin=1e-5) plt.savefig('bg_greyed.pdf',bbox_inches='tight') plt.close()
build_custom_continuous_cmap()¶
- sctriangulate.colors.build_custom_continuous_cmap(*rgb_list)[source]¶
Generating any custom continuous colormap, user should supply a list of (R,G,B) color taking the value from [0,255], because this is the format the adobe color will output for you.
Examples:
test_cmap = build_custom_continuous_cmap([64,57,144],[112,198,162],[230,241,146],[253,219,127],[244,109,69],[169,23,69]) fig,ax = plt.subplots() fig.colorbar(cm.ScalarMappable(norm=colors.Normalize(),cmap=diverge_cmap),ax=ax)
build_custom_divergent_cmap()¶
- sctriangulate.colors.build_custom_divergent_cmap(hex_left, hex_right)[source]¶
User supplies two arbitrary hex code for the vmin and vmax color values, then it will build a divergent cmap centers at pure white.
Examples:
diverge_cmap = build_custom_divergent_cmap('#21EBDB','#F0AA5F') fig,ax = plt.subplots() fig.colorbar(cm.ScalarMappable(norm=colors.Normalize(),cmap=diverge_cmap),ax=ax)
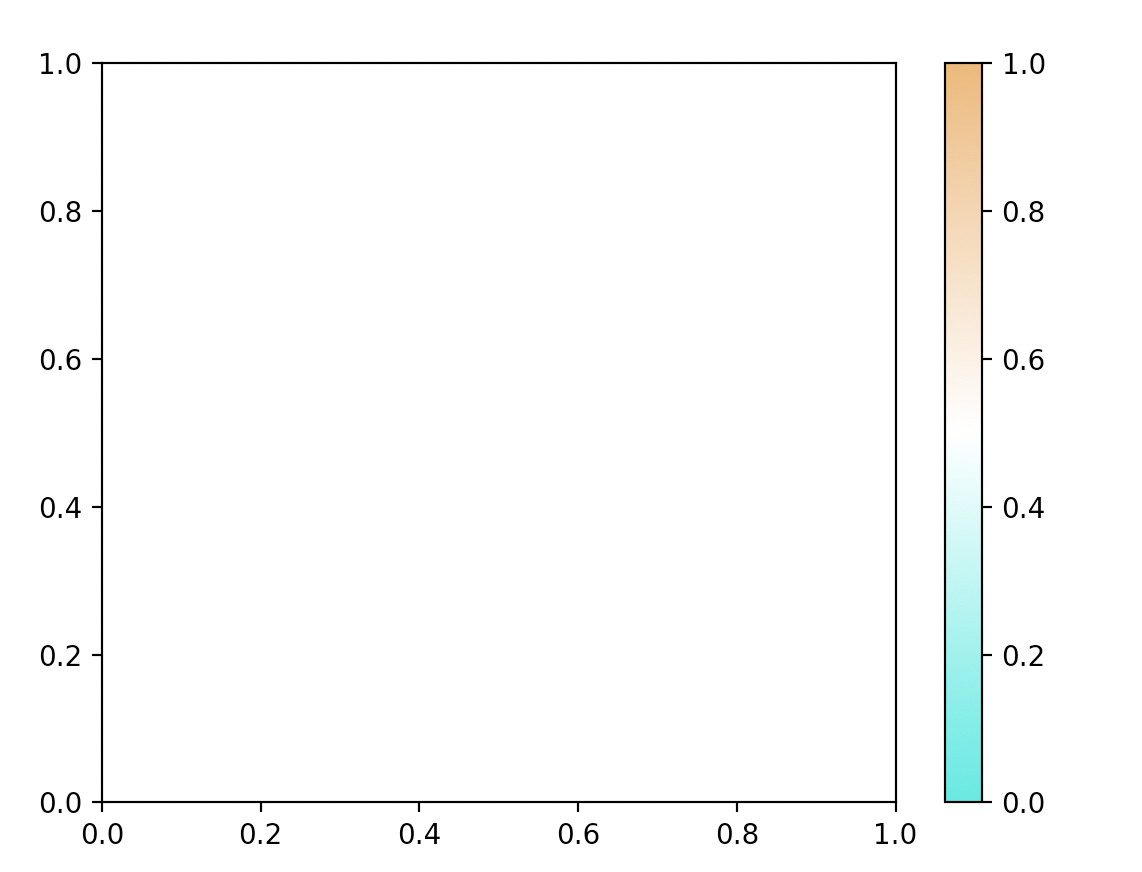
retrieve_pretty_cmap()¶
- sctriangulate.colors.retrieve_pretty_cmap(name)[source]¶
retrieve pretty customized colormap
- Parameters
name – string, valid value ‘altanalyze’, ‘shap’, ‘scphere’
- Returns
cmap object
Examples:
generate_gradient(cmap=retrieve_pretty_cmap('shap'),name='shap') generate_gradient(cmap=retrieve_pretty_cmap('altanalyze'),name='altanalyze') generate_gradient(cmap=retrieve_pretty_cmap('scphere'),name='scphere')

retrieve_pretty_colors()¶
- sctriangulate.colors.retrieve_pretty_colors(name)[source]¶
retrieve pretty customized colors (discrete)
- Parameters
name – string, valid value ‘icgs2’, ‘shap’
- Returns
list, each item is hex code
Examples:
generate_block(color_list = retrieve_pretty_colors('icgs2'),name='icgs2') generate_block(color_list = retrieve_pretty_colors('shap'),name='shap')
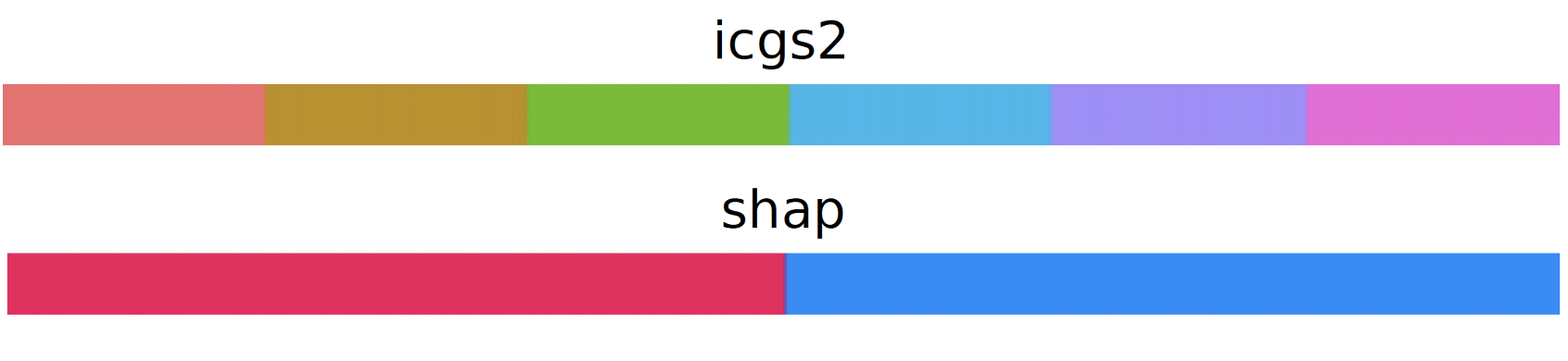
generate_block()¶
Spatial (Experimental)¶
read_spatial_data¶
- sctriangulate.spatial.read_spatial_data(mode_count='mtx', mode_spatial='visium', mtx_folder=None, txt_file=None, txt_file_sep=',', tmp_mtx_folder=None, spatial_library_id=None, spatial_folder=None, spatial_coord=None, spatial_coord_sep=',', coord_columns=None, spatial_images=None, spatial_scalefactors=None, **kwargs)[source]¶
read the spatial data into the memory as adata
- Parameters
mode_count –
string, how the spatial count data is present:
mtx: a folder where inputs are represented in mtx format
large_txt: a large txt file, usually means exceed 2GB
small_txt: a small txt file, usually means below 2GB
mode_spatial –
string, how the spatial images and associated files are present
visium: spatial location and images are laid out as visium format from spaceranger
generic: more generic format where the users have to supply the file path to find the location and images
Depending on how the mode_count is set, additional paramters need to be set for reading the count data
- Parameters
mtx_folder – string, if mode_count == ‘mtx’, specify the folder name
txt_file_sep – string, if mode_count == ‘small_txt’ for ‘large_txt’, it can be either common or tab or others
txt_file – string, if mode_count == ‘small_txt’ for ‘large_txt’, specify the txt path
tmp_mtx_folder – string, if mode_count == ‘large_txt’, specify the folder where the intermediate mtx folder will be created
Depending on how the mode_spatial is set, addtiional parameters need to be set for reading the spatial data
- Parameters
spatial_library_id – string, when necessary, specify the library_id for the spatial slide
spatial_folder – string, if mode_spatial == ‘visium’, specifiy the folder name, when mode_spatial==’visium’
spatial_coord – string, the path to which we have spatial coords for each barcode, when mode_spatial==’generic’
spatial_coord_sep – string, it can be ‘ ‘ or ‘,’ or other delimiters, when mode_spatial==’generic’
coord_columns – list, the columns you need to transfer from
spatial_coord, when mode_spatial ==’generic’spatial_images – dict, {‘hires’:path_to_image}, it can be None, when mode_spatial ==’generic’
spatial_scalefactors – dict, {‘tissue_hires_scalef’:0.17,’tissue_lowres_scalef’:0.05,’fiducial_diameter_fullres’:144,’spot_diameter_fullres’:89}, it can be None, when mode_spatial==’generic’
Depending on how the mode_count is set, additional parameters can be passed to the underlying preprocessing functions to read in the data
- Parameters
kwargs – optional keyword arguments will be passed to the function handling how the input count will be read
Examples:
id_ = '1160920F' adata_spatial = read_spatial_data(mtx_folder='filtered_count_matrices/{}_filtered_count_matrix'.format(id_), spatial_folder='filtered_count_matrices/{}_filtered_count_matrix/{}_spatial'.format(id_,id_), spatial_library_id=id_,feature='features.tsv')
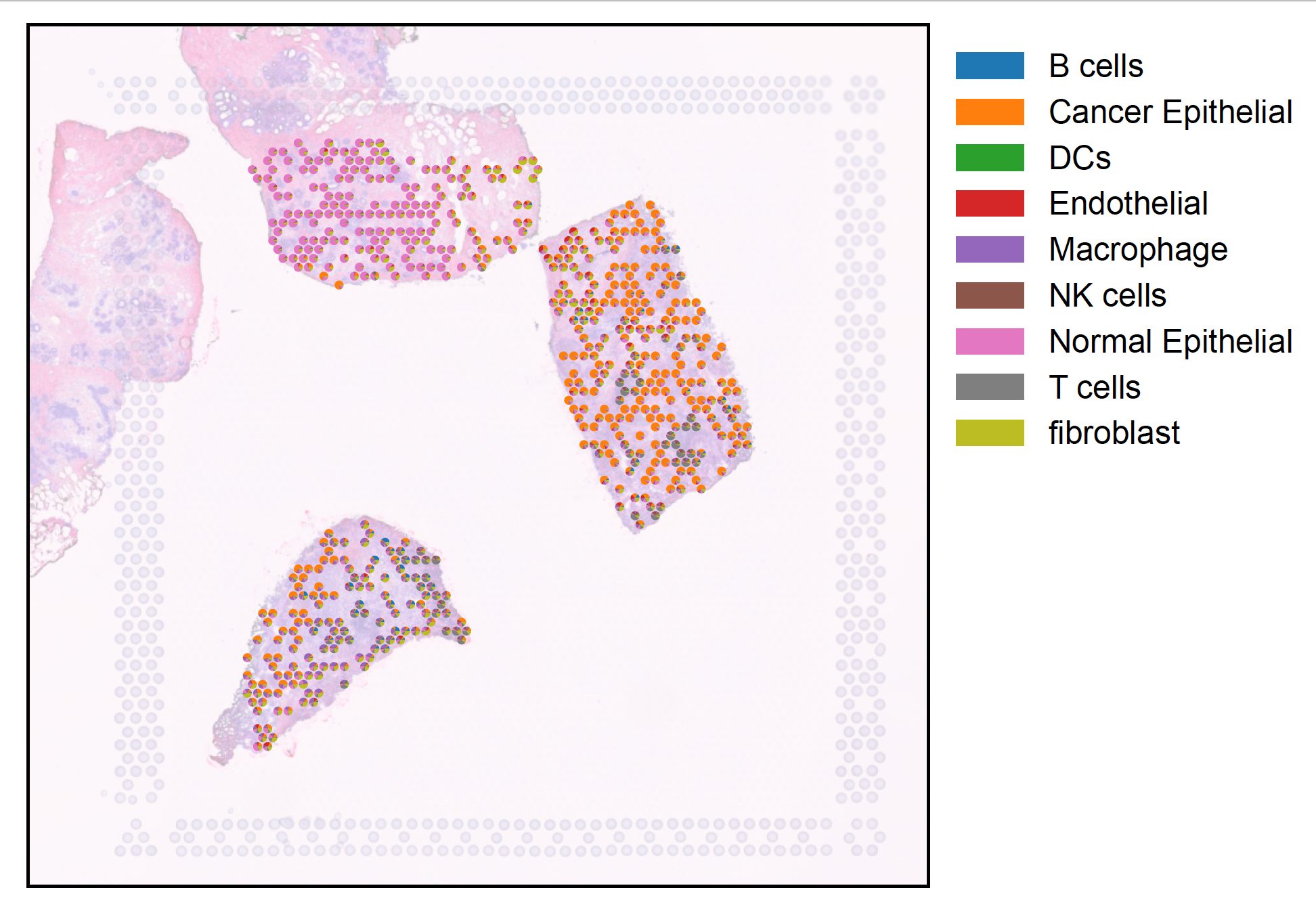
plot_deconvolution¶
- sctriangulate.spatial.plot_deconvolution(adata, decon, fraction=0.1, size=0.5, library_id=None, alpha=0.5, outdir='.')[source]¶
Visualize the deconvolution result as pie chart, serving as a complement function on top of scanpy.pl.spatial where each dot now is a pie chart
- Parameters
adata – AnnData, requires img, scale_factor, spot_diameter in the canonical slot based on squidpy convention, you can get it either using squidpy.read_visium or sctriangulate.spatial.read_spatial_data
decon – A dataframe whose index is spot barcode, column is the cell types, value represents cell type proportion (each row sums to 1)
fraction – float, plot that fraction of spot on the image, default is 0.1
size – float, default is 0.5, adjust it based on visual effect
library_id – None or string, if None, automatically extract from adata.uns[‘spatial’], or particularly specify
alpha – float, default is 0.5, the transparency of the underlying image
outdir – string, the output dir for saved image
Example:
decon = pd.read_csv('inputs/decon_results_A/cell2location_prop_processed.txt',index_col=0,sep=' ') plot_deconvolution(adata_spatial,decon,fraction=0.5,alpha=0.3)
cluster_level_spatial_stability¶
- sctriangulate.spatial.cluster_level_spatial_stability(adata, key, method, neighbor_key='spatial_distances', sparse=True, coord_type='generic', n_neighs=6, radius=None, delaunay=False)[source]¶
derive optional stability score in the context of spatial transcriptomics
- Parameters
adata – the Anndata
key – string, the column in obs to derive cluster-level stability score
method –
string, which score, support tbe following:
degree_centrality
closeness_centrality
average_clustering
spread
assortativity
number_connected_components
neighbor_key – string, which obsm key to use for neighbor adjancency, default is spatial_distances
sparse – boolean, whether the adjancency matrix is sparse or not, default is True
coord_type – string, default is generic, passed to sq.gr.spatial_neighbors()
n_neighs – int, default is 6, passed to sq.gr.spatial_neighbors()
radius – float or None (default), passed to sq.gr.spatial_neighbors()
delaunay – boolean, default is False, whether to use delaunay for spatial graph, passed to sq.gr.spatial_neighbors()
Examples:
cluster_level_spatial_stability(adata,'cluster',method='centrality') cluster_level_spatial_stability(adata,'cluster',method='spread') cluster_level_spatial_stability(adata,'cluster',method='assortativity',neighbor_key='spatial_distances',sparse=True) cluster_level_spatial_stability(adata,'cluster',method='number_connected_components',neighbor_key='spatial_distances',sparse=True)
create_spatial_features¶
- sctriangulate.spatial.create_spatial_features(adata, mode, coord_type='generic', n_neighs=6, n_rings=1, radius=None, delaunay=False, sparse=True, library_id=None, img_key='hires', sf_key='tissue_hires_scalef', sd_key='spot_diameter_fullres', feature_types=['summary', 'texture', 'histogram'], feature_added_kwargs=[{}, {}, {}], segmentation_feature=False, segmentation_method='watershed')[source]¶
Extract spatial features (including spatial coordinates, spatial neighbor graph, spatial image)
- Parameters
adata – the adata to extract features from
mode –
string, support:
coordinate: feature derived from pure spatial coordinates
graph_importance: feature derived from the spatial neighbor graph
tissue_images: feature derived from assciated tissue images (H&E, fluorescent)
coord_type – string, default is generic, passed to sq.gr.spatial_neighbors()
n_neighs – int, default is 6, passed to sq.gr.spatial_neighbors()
n_rings – int, default is 1, passed to sq.gr.spatial_neighbors()
radius – float or None (default), passed to sq.gr.spatial_neighbors()
delaunay – boolean, default is False, whether to use delaunay for spatial graph, passed to sq.gr.spatial_neighbors()
library_id – string or None, when choosing tissue_images, need to retrieve the image from adata.uns
img_key – string, when choosing tissue_images, need to retrieve the image from adata.uns
sf_key – string, when choosing tissue_images, we need to associate image pixel to the spot location, which relies on scalefactor
sd_key – the string, the key of spot_diameter_fullres in adata.uns[‘spatial’][libarary_id][‘scalefactorf’]
feature_types – list, default including summary, texture and histogram
feature_added_kwargs – nested list, each element is a dict, containing additional keyword arguments being passed to each feature function in feature_types
segmentation_feature – boolean, whether to extract segmentation feature or not, default is False
segmentation_method – string the segmentation method to use, default is ‘watershed’
Examples:
# derive feature spatial_adata_image = create_spatial_features(adata_spatial,mode='tissue_image',library_id='CID44971',img_key='hires',segmentation_feature=True) # derive spatial clusters sc.pp.neighbors(spatial_adata_image) sc.tl.leiden(spatial_adata_image) spatial_adata_image.uns['spatial'] = adata_spatial.uns['spatial'] spatial_adata_image.obsm['spatial'] = adata_spatial.obsm['spatial'] sc.pl.spatial(spatial_adata_image,color='leiden')
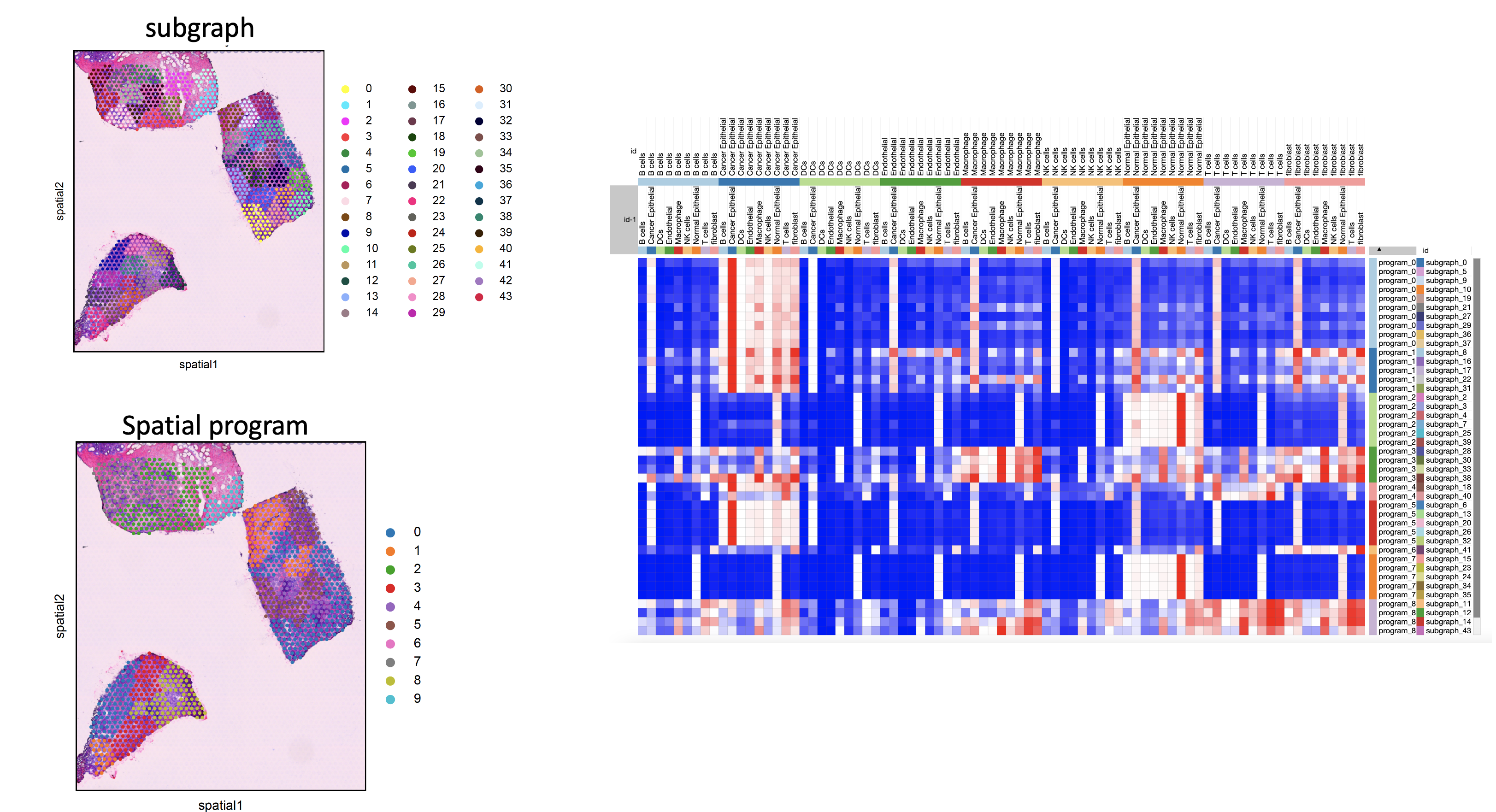
identify_ecosystem¶
- sctriangulate.spatial.identify_ecosystem(adata_spatial, coord_type='grid', n_neighbors=6, n_rings=1, include_self=True, resolution=1, save=True, outdir='.', legend_loc='right margin')[source]¶
Ecosystem means the frequent interaction within one cell type, or across multiple cell types. Examples include:
A ecosytem within which macrophage interact with macrophage
A ecosytem where stroma cells encapsulating tumor tissue
A ecosytem where T cell and B cell co-occur
This function is inspired by the recurrent cellular neighborhood analysis described in The Spatial Landscape of Progression and Immunoediting in Primary Melanoma at Single-Cell Resolution.
- Parameters
adata_spatial – Anndata, follow the squidpy convention in terms of the slot where img, scale_factor should go into, further more, please put decon dataframe into adata_spatial.obsm[‘decon’]
coord_type – either grid or generic, passed to sq.gr.spatial_neighbors function
n_neighbors – int, default is 6, passed to sq.gr.spatial_neighbors function
n_rings – int, default is 1, passed to sq.gr.spatial_neighbors function
include_self – boolean, when counting neighbors, whether or not including the spot itself
resolution – float, default is 1, this will be passed to leiden algorithm
save – boolean, whether to save the plot or not
outdir – string, default is ‘.’, output directory
legend_loc – string, passed to sq.pl.spatial, default is right margin
Example:
adata_spatial.obsm['decon'] = decon.loc[adata_spatial.obs_names,:] adata_neigh = identify_ecosystem(adata_spatial,n_rings=2) sc.pl.spatial(adata_neigh,color='leiden',groups=['6'],alpha_img=0.2)
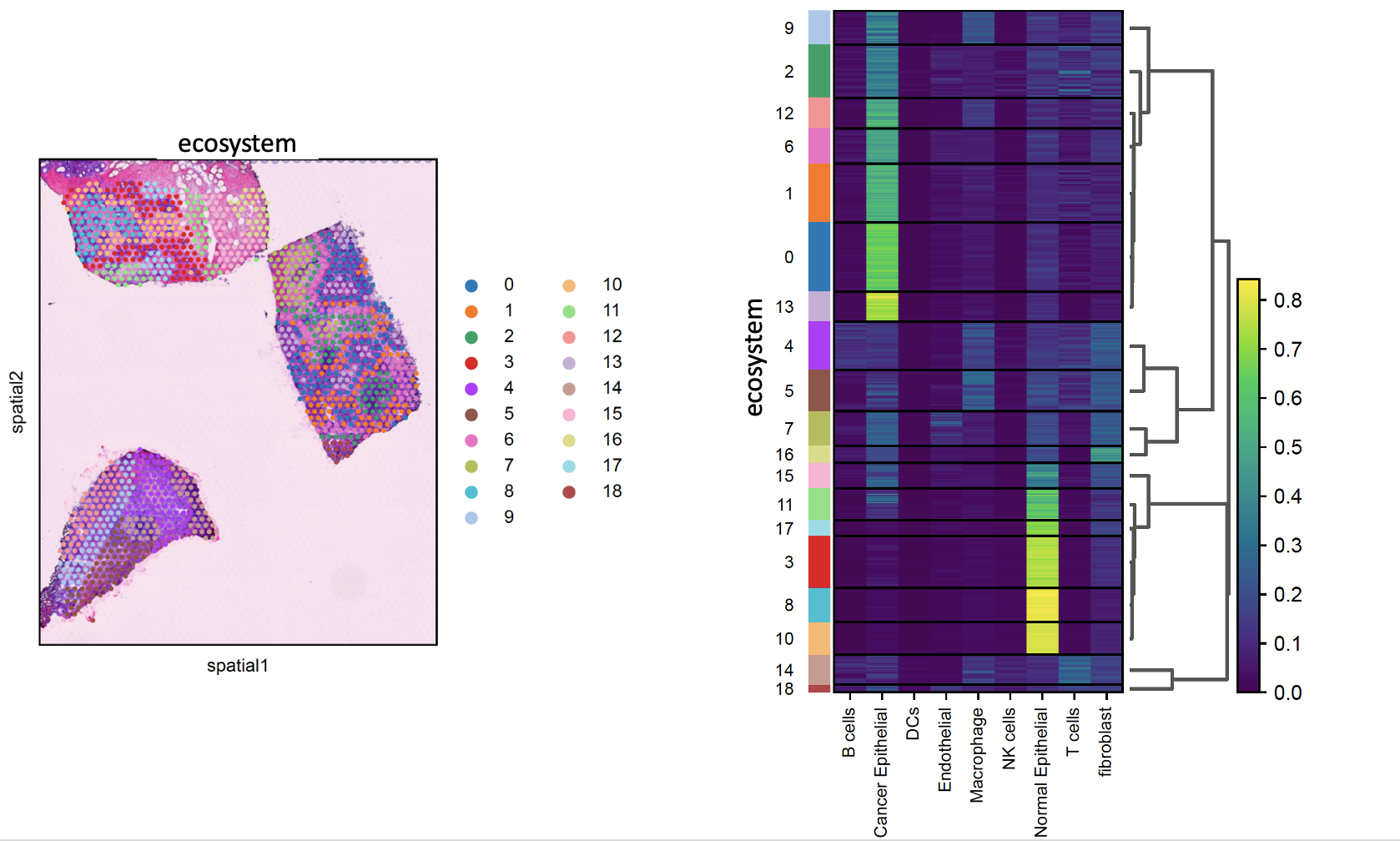
identity_spatial_program¶
- sctriangulate.spatial.identify_spatial_program(adata_spatial, coord_type='grid', n_neighbors=6, n_rings=1, spatial_community_resolution=8, spatial_community_min_cells=10, plot=True, outdir='.', mode='spot_cell_type_proportion', n_program=10)[source]¶
The most important analysis is to define spatial program, meaning spatial region that are proximal and somehow transcriptomically similar. This function provide a very flexible way to define spatial program by first derive spatial community solely based on spatial coordinates, then we merge spatial community into spatial cluster/structure such that spatial communities sharing similar gene expression or cell phenotype profile will be clustered tegether, the resultant spatial cluster represents the biologically meaningful multicellular spatial structure, it is inspired by Figure 4 in this paper.
- Parameters
adata_spatial – the Anndata with spatial coordinate
coord_type – either grid or generic, passed to sq.gr.spatial_neighbors function
n_neighbors – int, default is 6, passed to sq.gr.spatial_neighbors function
n_rings – int, default is 1, passed to sq.gr.spatial_neighbors function
spatial_community_resolution – float, default is 8, passed to nx.algorithms.community.greedy_modularity_communities, high value means smaller clusters
spatial_community_min_cells – int, the minimum number of cells/spots that a spatial community needs to have
plot – boolean, whether to plot spatial community and spatial cluster
outdir – string, the path to the output dir
mode – string, which transcriptomical profile to consider, it can be ‘spot_cell_type_proportion’ coming out of spatial deconvolution methods or spot gene expression
n_program – int, the number of spatial program you want, it is passed to hierarchical clustering algorithm
- Return df_subgragh
dataFrame, readily input for Broad Morpheus online heatmap generation tool.
Examples:
adata_spatial.obsm['decon'] = decon.loc[adata_spatial.obs_names,:] df_subgraph = identify_spatial_program(adata_spatial,n_rings=1)